TMEPAI(prostate transmembrane protein,androgen induced 1,遺伝子名PMEPA1)は,前立腺細胞でアンドロゲンやその誘導体によって発現が亢進するタンパク質として同定され1),別名PMEPA1, STAG1, ERG1.2, N4wbp4とも呼ばれている(本稿ではTMEPAIと統一する).筆者らは,DNAマイクロアレイを用いた網羅的スクリーニングにより,NMuMG細胞でトランスフォーミング増殖因子β(TGF-β)によって誘導される遺伝子としてTMEPAIを同定し,さらのTMEPAIがTGF-βの直接の標的遺伝子であることを明らかにした2, 3).ほぼ同時期に上皮増殖因子(EGF)などの増殖シグナルや変異型p53によっても誘導されることがわかり,さまざまな刺激によって誘導される遺伝子の一つとして認識されるようになった4–6).筆者らは,TMEPAIがTGF-βシグナルの細胞内シグナル伝達分子であるSmad2とSmad3に結合して,TGF-βシグナルを抑制することを見いだした.一方,TMEPAIと相同性の高いC18ORF1[遺伝子名low density lipoprotein receptor class A domain containing 4(LDLRAD4),本稿ではC18ORF1と統一]もTMEPAIと同様にTGF-βシグナルを抑制することがわかり,筆者らはこれら二つの分子をTMEPAIファミリーと呼んでいる3, 7).最近TMEPAIがTGF-β I型受容体[activin receptor-like kinase 5(ALK5)]の分解8)やオートファジー形成9)にも関与していることがわかってきている.加えて,アンドロゲン受容体(AR)シグナルを制御する分子としての役割も唱えられており10–13),TMEPAIはマルチシグナル制御分子として,細胞の恒常性維持に関与しうる.また,腫瘍組織などでTMEPAIの発現異常に関する報告もあり,TMEPAIと腫瘍進展との関連も注目されている.
本稿では,TGF-βシグナルについて簡単に述べた後,TMEPAIファミリーによるTGF-βシグナル調節を中心にTMEPAIファミリーのマルチシグナル制御因子としての機能および疾患との関係についての最近の話題を紹介する.
1)TGF-βファミリー
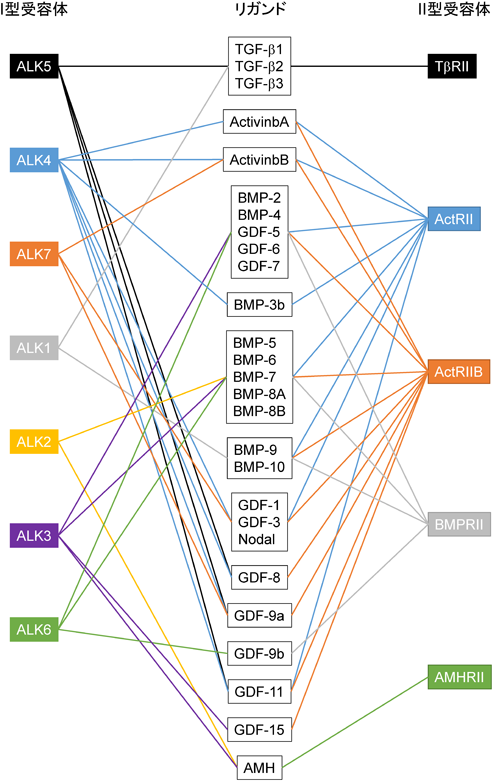
TGF-βは,タンパク質の構造が類似した他のサイトカインと共にTGF-βファミリーと呼ばれており,ヒトゲノム中に33種類の類縁タンパク質が存在している.歴史的には,腫瘍細胞培養上清中に発見されたTGF-βが軟寒天培地で正常繊維芽細胞の足場非依存性増殖を誘導したことから,発見当初は腫瘍増殖を促進するサイトカインであると考えられていた.しかしその後,TGF-βは細胞増殖促進よりも上皮細胞や血球細胞に対する細胞増殖抑制能が広く知られるようになった.加えて,血球分化を促進するアクチビン(activin)や骨・軟骨形成に関与するBMP(bone morphogenetic protein)などがファミリー分子であることがわかり,現在アクチビンやBMPとともに,TGF-βはTGF-βファミリーを形成している14, 15)(図1).TGF-βは,細胞増殖抑制作用以外に細胞分化,アポトーシス,遊走能,細胞外マトリックスタンパク質産生,血管新生,免疫制御,がん化促進など,多様な生理活性を有している.前がん状態において,TGF-βは細胞増殖抑制作用によりがん細胞の増殖を抑制するが,悪性化したがん細胞では,その細胞増殖抑制作用が消失する一方,細胞運動能亢進や細胞外マトリックス分解酵素誘導などの作用により,がん細胞の浸潤能を亢進させる.浸潤能を獲得したがん細胞はTGF-βを分泌するようになり,オートクリンおよびパラクリン反応によってさらに細胞運動を活性化する.加えて,免疫細胞に属する細胞傷害性T細胞やNK細胞の増殖を抑制することでがん細胞を標的とする免疫監視システムからがん細胞を回避させ,血管新生を亢進することで酸素や栄養素を獲得できるがん微小環境作りに一役を買っている.したがってTGF-βは,がん化の初期ではがん進展を抑制するが,悪性化したがん細胞ではがん化を亢進するという二面性を有している16, 17).
2)TGF-β受容体
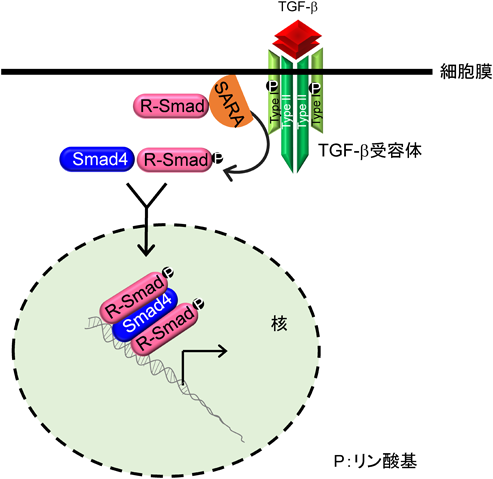
TGF-βファミリー分子に対する受容体として,7種類のI型受容体,5種類のII型受容体が存在し,それらすべての受容体は細胞内領域にセリン/トレオニンキナーゼドメインを持つ.ジスルフィド結合により二量体を形成しているTGF-βは,2分子のTGF-β I型受容体と2分子のTGF-β II型受容体からなるヘテロ四量体に結合する(図2).リガンドがTGF-β II型受容体に結合すると,TGF-β II型受容体キナーゼはTGF-β I型受容体の膜貫通領域直下に存在するGS領域と呼ばれるグリシンとセリンに富んだドメインのセリンとトレオニン残基をリン酸化し,I型受容体内で不活性化されていたセリン/トレオニンキナーゼ酵素活性が出現する.TGF-βと同様にアクチビンは,アクチビンI型受容体(ALK4)とアクチビンII型受容体(ActRIIまたはActRIIB)が複合体を形成し,BMPの場合BMPI型受容体(ALK2, 3, 6)とBMP II型受容体(ActRII,ActRIIBまたはBMPRII)が複合体を形成する.活性化したI型受容体キナーゼはR-Smad(receptor-regulated Smad)タンパク質のC末端に存在する2個のセリン残基をリン酸化する.TGF-βやアクチビン刺激でリン酸化されるR-Smad(Smad2とSmad3)をAR-Smad(TGF-β/activin R-Smad),BMP刺激でリン酸化を受けるR-Smad(Smad1, Smad5, Smad8)をBR-Smad(BMP R-Smad)と呼ぶ.リン酸化を受けた2分子のR-SmadはSmad4とヘテロ三量体を形成し,核内に移行することで標的遺伝子の転写を調節する17, 18)(図2).
3)Smad依存的TGF-βシグナル
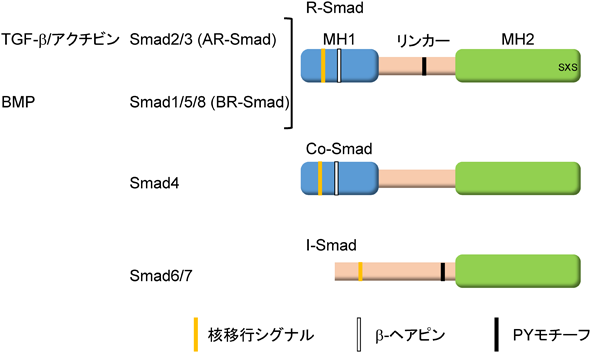
Smadは哺乳動物で8種類存在している.TGF-β I型受容体であるALK5は,ALK4とALK7とともにAR-Smadのリン酸化を触媒する.一方,BR-SmadはBMP I型受容体であるALK2, ALK3, ALK6およびALK1によってリン酸化される.リン酸化R-Smadと複合体を形成するSmad4をcommon partner Smadまたはcommon mediator Smad(Co-Smad)と呼び,その他にSmad6とSmad7から構成されているinhibitory Smad(I-Smad)が知られており,I-SmadはTGF-βファミリーシグナルを抑制する.
Smadはその構造上の特徴から,三つの領域に分けられている.N末端側はMH(mad homology)1領域,C末端側はMH2領域,その間はSmad間で相同性が低いためリンカー領域と呼ばれる(図3).MH1領域はI-Smadを除くすべてのSmad間で高く保存されている.リンカー領域には,さまざまなシグナル系によりリン酸化される部位やE3ユビキチンリガーゼと結合するPYモチーフが存在し,Smadの機能や安定性に影響を与えることが知られている.R-SmadとCo-SmadはMH1領域に核移行シグナルおよびDNAとの結合に重要なβ-ヘアピン構造を持つ(Smad2は,β-ヘアピン構造に隣接してSmad2固有のアミノ酸配列が挿入されているため,DNAに結合できない).MH2領域はI型受容体や転写因子との結合に重要な領域であることが知られている(図3).リン酸化されたR-Smadは,Co-SmadであるSmad4と三者複合体を形成して核に移行し,標的遺伝子のプロモーターのCAG Aまたはそれに類似した固有のDNA結合配列(Smad binding element:SBE)に直接または他の転写因子を介して間接的に結合し,その転写を制御している19, 20)(図2).Smad自身はDNAへの親和性が低いため,他の転写因子と会合して間接的にDNAに結合する場合もある21).Smadタンパク質はTGF-β I型受容体によるR-SmadのC末端のリン酸化以外にSmad自身の分解を促進するポリユビキチン化,Smadの機能を修飾するモノユビキチン化やSUMO化,アセチル化やADPリボシル化などを受け,機能は調節されている.Smadの翻訳後修飾については,他の論文に詳細が記載されている20, 22, 23).
4)Smad非依存的TGF-βシグナル
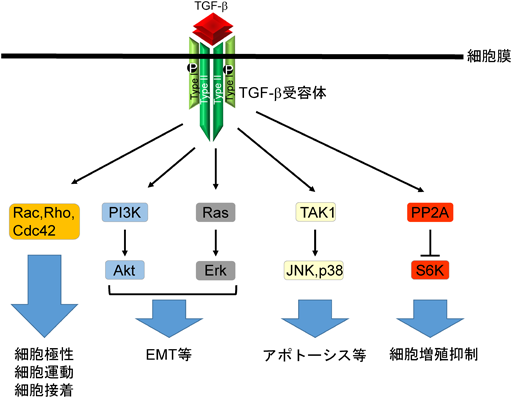
SmadシグナルはTGF-βシグナル伝達に重要な役割を果たしているが,活性化されたTGF-β受容体からSmad非依存的シグナルも伝えられることが知られ,場合によってはSmadシグナルの活性を補助している2).Smad非依存的シグナルとしては,MAPキナーゼ経路のErk, JNK, p38やPI3キナーゼ,PP2A,ならびにRhoシグナルが有名である.これらのSmad非依存的シグナル系は,Smad依存的シグナルと異なり,すべての細胞が有するシグナル経路ではなく,細胞の種類やその細胞が置かれた環境に固有の経路である24, 25)(図4).
5)TGF-βシグナルのネガティブフィードバック機構
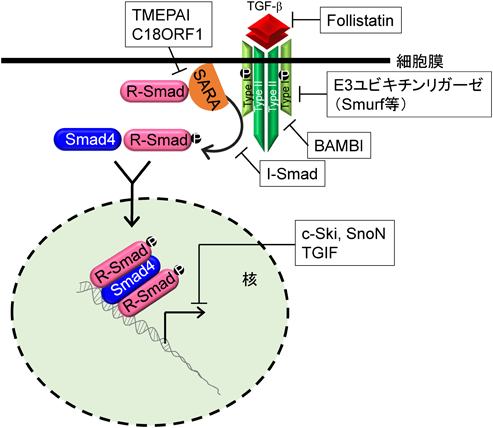
細胞は細胞外から核内までのステップで過度のTGF-βシグナルを抑制するシステムを有しており,このシステムの破綻が疾病につながると考えられている.たとえば,細胞外ではTGF-βファミリーであるアクチビンをトラップするfollistatin,細胞膜ではデコイ受容体であるBAMBI(BMP and activin membrane-bound inhibitor homolog),細胞質ではI-SmadやさまざまなE3ユビキチンリガーゼ,そして核内では転写抑制共役因子c-Ski, SnoN, TGIF(TGF-β-induced factor homeobox)などである(図5).これらのTGF-βシグナルを抑制する分子の中には,TGF-βシグナルの直接の標的遺伝子も含まれる.特にSmad7やSnoNに加え本稿で取り上げているTMEPAIが有名であり,これらの分子はTGF-βシグナルのネガティブフィードバックに関与している.TGF-βファミリーによって誘導されるSmad7は,TGF-β刺激により細胞内局在が核から細胞質へと変化し26),E3ユビキチンリガーゼであるSmurfや脱リン酸化酵素であるPP2Cと複合体を形成する.Smad7はTGF-β I型受容体であるALK5と直接結合できるため,Smad7を介してSmurfはALK5をポリユビキチン化しプロテオソーム依存的分解を行い,PP2CはALK5の脱リン酸化を行うことでALK5セリン/トレオニンキナーゼを不活化状態に導きTGF-βシグナルを抑制する.興味ある点として,Smad7はTGF-βファミリーI型受容体すべてのセリン/トレオニンキナーゼ活性を抑制する.一方,I-Smadに属するSmad6もTGF-βやBMPによって誘導されるが,Smad7と異なりBMP I型受容体からのシグナルを特異的に抑制する.Salt-inducible kinase(SIK)もTGF-βシグナルにより誘導され,Smad7と複合体を形成することでTGF-β I型受容体の分解を促進する.SnoNはDNA結合活性を有していないが,核内でリン酸化R-Smad/Smad4複合体に結合することで転写抑制共役因子であるN-CoR, mSin3A, CtBPをリクルートし,転写活性化共役因子であるp300/CBPを転写複合体から解離させ,転写を抑制する17)(図5).
1)TMEPAIの構造
TMEPAIは,アンドロゲンで誘導される遺伝子産物として単離されたI型膜貫通タンパク質である1).その後,筆者らがTGF-βによって誘導されるタンパク質であることを報告した2, 3).また,EGF,変異型p53, wntシグナル,低酸素刺激で誘導されるタンパク質でもある3–7, 27, 28).TMEPAIは約40アミノ酸から構成され,保存された2つのトリプトファン残基が存在するWWドメインと結合できるPYモチーフを2個有しており,NEDD4結合タンパク質としても見いだされ10),ヒトにおいて5種類のアイソフォームが存在している(図6A).最も分子量が大きいアイソフォームは287アミノ酸残基からなっており,その他の4種類のアイソフォームはN末端側に一部の相異がある以外,膜貫通領域や細胞質領域は同一である.細胞内の特徴的な領域として,上述した二つのPYモチーフとその間に位置するSIMドメイン(Smad interactingモチーフ,詳細は後述)がある(図6A).
2)C18ORF1の構造
TMEPAIに類似したアミノ酸配列を持つC18ORF1には,8種類のアイソフォームが存在する.TMEPAIと同様にN末端領域の違いからC18ORF1α, C18ORF1β, C18ORF1γ, C18ORF1δ, C18ORF1εがあり,C18ORF1α, C18ORF1β, C18ORF1γにはそれぞれ膜貫通領域直下に18アミノ酸を欠失したアイソフォームも存在している29)(図6B).膜貫通領域よりC末端側では,18アミノ酸欠失以外はC18ORF1アイソフォーム間でまったく同じアミノ酸配列である.C18ORF1α1が最も長く,306アミノ酸からなり,TMEPAIと同様に二つのPYモチーフとSIMドメインを有している.C18ORF1αアイソフォームのN末端領域にLDLRAD4に類似した構造があるが,実際にLDLなどのリポタンパク質と結合するという報告はない(図6B).
1)TGF-βシグナルによるTMEPAI遺伝子発現制御
TGF-βによる誘導に必要な転写制御領域はTMEPAI遺伝子の転写開始点上流約2 kbまでにはなく,第1イントロン内の+1037~+1294の領域に存在した.この領域には5個のSBEと4個のTBE(TCF/LEF binding element)が存在するが,そのうち5´側の3個のSBEと1個のTBE[筆者らはTGF-β-responsive TCF7L2-binding element(TTE)と名づけた]がTMEPAI遺伝子発現に重要な転写制御配列である.TMEPAI遺伝子発現制御には,Smad3とSmad4から構成される複合体のみならず,TCF/LEFファミリーのTCF7L2とElk-1も協調して働いている27, 30).Fournierらも,TMEPAI遺伝子の転写開始点から上流約3.7 kbまでを用いてTGF-β応答に必要な転写制御配列の同定を行ったが,この領域内に存在するすべてのSBEに変異を導入してもTGF-β感受性に変化は認められず,筆者らの結果を裏づけるものとなっている31).
2)SP1によるTMEPAI遺伝子発現制御
SP1は,GC-rich領域に結合し,ハウスキーピング遺伝子のプロモーターに高頻度で結合する転写因子である.TMEPAI遺伝子の転写開始点から上流−298の間に2個存在するSP1結合配列が,TMEPAI遺伝子の基本転写活性に影響を与えている32).
3)WntシグナルによるTMEPAI遺伝子発現制御
自然発症消化管がんモデルであるApcMIN/+マウスの消化管ポリープにおいて,TMEPAIの発現が亢進していることより,WntシグナルがTMEPAI遺伝子発現を誘導すると推測された3, 27, 28).実際,Wntシグナルを活性化する塩化リチウム刺激やβ-カテニン遺伝子の強制発現により,TMEPAI遺伝子が活性化された27).古典的Wntシグナルとしてβ-カテニンが核内でTCF/LEFファミリーと結合して,転写を促進することが広く知られている.TCF/LEFファミリーは4種類の分子からなり,その中でTCF7L1とTCF7L2はTMEPAI遺伝子発現を促進したが,LEF1とTCF7はできなかった.TCF7L1とTCF7L2は,LEF1とTCF7に比較してDNA結合領域であるHMG DBD(high mobility group DNA binding domain)のC末端側が150アミノ酸程度長くなっており,TCF7L2のC末端の約150アミノ酸をLEF1やTCF7に融合したキメラタンパク質は,TMEPAI遺伝子発現を促進させることができた.このC末端側にはSmad3との結合領域があり,TMEPAI遺伝子プロモーター上でSmad3がTCF7L2やTCF7L1と複合体を形成することで,TMEPAI遺伝子発現が活性化されると考えられる33).
4)TMEPAI遺伝子発現のエピジェネティクス制御
TMEPAI遺伝子は前立腺がんで高メチル化を受けており,前立腺がん細胞をメチル化酵素阻害剤で処理すると,TMEPAI遺伝子の発現が上昇する34).特にTMEPAI遺伝子の第1イントロン内の制限酵素HhaIで切断されるGCG C配列が,前立腺がんで高頻度にメチル化されていた.この配列がメチル化されている場合にTMEPAI遺伝子発現が抑制され,DNAメチル化酵素阻害剤によりその発現が回復した35).
5)その他のTMEPAI遺伝子発現制御
in silico解析によりTMEPAI遺伝子上流に2か所のAR結合領域が存在し,ChIPアッセイによりARが結合することが報告された36).しかしながら,TMEPAI遺伝子の転写活性化への関与についてわかっていない.その他,TMEPAI遺伝子はEGFなどの細胞増殖因子やp53変異体(121番目のセリンがフェニルアラニンに置換),低酸素刺激によって発現が誘導されるが4–6),その詳細な転写調節機構についてもいまだ不明のままである.
5. TMEPAIファミリーによるTGF-βシグナル抑制機構
1)TMEPAIによるTGF-βシグナル抑制機構
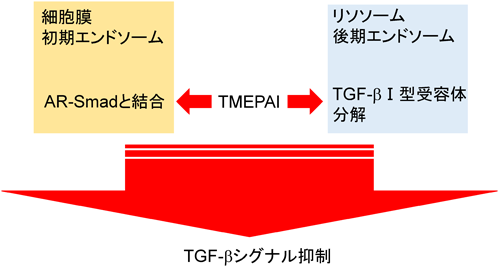
TGF-β標的遺伝子の中には,Smad7をはじめとしてTGF-βシグナルのネガティブフィードバックループに関与する遺伝子がある37, 38).TMEPAIはTGF-β依存的なAR-Smadのリン酸化を抑制したが,BMPシグナル下流のBR-Smadのリン酸化を抑制しなかった.またTMEPAIは膜貫通領域を有しているが,TGF-βと直接結合することはできなかった.この事実は膜貫通領域の一部が欠損し細胞質内に局在するTMEPAIアイソフォームc(本文中ではvariant 4/5)も,TGF-βシグナルを抑制できる事実と一致する.またTMEPAIは,ALK5やTGF-β II型受容体とも結合することができなかった.一方,TMEPAIはリン酸化の有無に関わらずAR-Smadと結合することができた.転写因子milkやmixerが持つAR-Smad結合領域(SIMドメイン:PPNR)がTMEPAIの細胞内領域にも存在するが,これら4アミノ酸をすべてアラニンに置換したTMEPAI変異体はAR-Smadとの結合能を喪失した.逆にAR-SmadであるSmad2内のSIM結合領域中心に位置する368番目のトリプトファンをアラニンに置換したSmad2変異体もTMEPAIと結合できないことがわかり,TMEPAIのSIMドメインとSmad2の368番目のトリプトファンを含んだ領域が互いに結合する可能性が示唆された.以上のことから,TMEPAIはAR-Smadを捕捉することでTGF-β I型受容体がAR-Smadと結合することを阻害していることがわかった3).これまでの報告より,TGF-β I型受容体がTGF-β刺激時にAR-Smadと結合するには,足場タンパク質であるSARA(Smad anchor for receptor activation)の介在が必要である39).SARAは自身が持つFYVEドメインを介して初期エンドソーム膜上のリン脂質と結合しており40),さらにSBD(Smad binding domain)でAR-Smadを捕捉し,リン酸化された活性化TGF-β I型受容体セリン/トレオニンキナーゼにAR-Smadを提示する39).実際筆者らは,TMEPAIがSARAと競合してAR-Smadと結合することで,SARAがAR-SmadをTGF-β I型受容体に提示することを阻害することを明らかにした.加えてSARAとTMEPAIは初期エンドソームで共局在していた.カエル初期胚において,TGF-βと同じシグナル伝達系を持つアクチビンは背側中胚葉を誘導する.TMEPAIを初期胚に導入すると,頭部や尾部が欠損して背側が開いた表現型が観察され,背部中胚葉形成阻害が起こった.さらにカエル胚を用いたアニマルキャップアッセイでは,TMEPAIの発現を抑制するとアクチビンで誘導されるXbraやGscの発現が促進され,逆にTMEPAIを大量発現するとこれらの発現が抑制されることがわかった.同様に培養細胞を用いた実験においても,TMEPAIを大量発現させるとTGF-βシグナルで誘導される上皮間葉転換(EMT)が抑制され,逆にTMEPAIの発現を抑制するとEMTが亢進されることを見いだした.以上の結果から,TGF-βシグナルによって誘導されたTMEPAIはSARAと競合してAR-Smadと結合し,SARAによるTGF-β I型受容体キナーゼへのAR-Smadの提示を阻害することでTGF-βシグナルを抑制すると思われる3)(図5,図7).
最近,FournierらもTMEPAIがTGF-βシグナルを抑制することを報告しているが,彼らの報告では,TMEPAIが足場タンパク質となり,PYモチーフを介してHECTタイプのE3ユビキチンリガーゼ(NEDD4, NEDD4-2, AIP4, Smurf2)と結合し,SIMドメインを介してAR-Smadと結合し,両分子が接近することでTGF-βシグナルが抑制されると述べている.この抑制機構はポリユビキチン化依存的でもプロテオソーム依存的でもないと結論づけているが,詳細なメカニズムは不明のままである.加えて,彼らの実験では膜貫通ドメインを欠いたTMEPAIがTGF-βシグナル抑制活性を持たないと報告している点で筆者らの報告と相反している.筆者らの場合,細胞膜貫通ドメインを欠いたTMEPAIアイソフォームcはTGF-βシグナルを抑制することができたが,その抑制作用は野生型のTMEPAIに比較して弱かった.さらに彼らは前立腺がんの患者由来の腫瘍組織を解析し,TMEPAI発現が高い場合,ARシグナルとTGF-βシグナルが阻害されることで細胞増殖と骨転移が抑制され,患者の生存率が高まり,一方DNAのメチル化によりTMEPAI遺伝子発現が低くなると,TGF-βシグナル亢進に引き続く転移関連遺伝子群の発現が上昇することで骨転移が起こり,患者の生存率が低下した31).またBaiらはTMEPAIがリソソームや後期エンドソームに存在し,TGF-β I型受容体がTMEPAIによってリソソームにリクルートされ,タンパク質分解を受けることによりTGF-βシグナルが抑制されると報告している8).
このようにTMEPAIがTGF-βシグナルを抑制することは確かであるが,その作用機序についてはいくつかの異なる報告があり,混沌としている.最近TMEPAIの発現を抑制するとリソソーム膜が不安定化され,オートファジーが阻害されることも見いだされている9).TGF-βシグナルがオートファジー制御にも関与していることからも41–43),TMEPAIによるオートファジー制御における分子機構の解明が今後期待される.
2)C18ORF1によるTGF-βシグナル抑制機構
筆者らは,TMEPAIに相同性のある分子を検索し,C18ORF1を同定した.C18ORF1とTMEPAIはN末端側の細胞外領域はまったく相同性を持たないが,アミノ酸配列で膜貫通領域は75%,細胞内領域は67%のアミノ酸相同性を持つ.図6Bに示したように,C18ORF1はSIM領域を持つためTMEPAIと同様にTGF-βシグナルを特異的に抑制するが,その抑制メカニズムもTMEPAIとまったく同様であった.ただし,C18ORF1はTMEPAIと異なり恒常的に発現しており,定常状態ではTGF-βシグナルに対するゲートキーパーとして作用すると考えられる.一方,C18ORF1のみで抑えることができない過度のTGF-βシグナルを細胞が受け取るとTMEPAIの発現が誘導され,C18ORF1と協同してTGF-βシグナルを抑制すると考えられる7)(図5).
1)TMEPAIによるARシグナル制御
AR陽性前立腺がんでは,TMEPAIは通常NEDD4を介してARのポリユビキチン化を促進することでARの発現を低下させ,細胞増殖を抑制する.しかしながら,TMEPAIの発現ががん化の過程で極端に低下すると,ARの発現亢進と細胞増殖促進(S期の細胞の割合増加)が認められる.したがって,TGF-βシグナルのみならずARシグナルもTMEPAIはネガティブフィードバック機構で阻害し,前立腺がんの進行を抑制すると考えられている10, 11).加えて,TMEPAI発現が抑制されたAR陽性前立腺がんでは,ARシグナルでNEDD4によるPTEN(phosphatase and tensin homolog deleted on chromosome 10)分解が促進されてPI3K/Aktシグナル経路が活性化され,がん増殖やがん進展が亢進する12)(図8A).このような報告と相反して,AR陰性の前立腺がんでは,TMEPAIの過剰発現がTGF-β標的遺伝子であるp21の発現抑制とc-Mycの発現亢進を導き,前立腺がんを進行させる13)(図8B).AR陽性前立腺がんとAR陰性前立腺がんでTMEPAIの果たす機能が異なることは,TMEPAIを標的とした抗がん剤開発を行う際に着目しなければいけない点であろう.
2)TMEPAIによる活性酸素産生に伴うEMT促進
Huらは,A549細胞においてTGF-β刺激によるEMTがTMEPAIの発現を抑制することで阻害されることを見いだし,TGF-βによって促進されるEMTにTMEPAIが必要であると報告している.そして,TMEPAIがTGF-β依存的に抑制されたフェリチン発現を改善することで活性酸素産生を増加させると同時に,EMTを抑制するIRS-1の発現を抑制して,EMTを促進させると結論づけている44).しかしながら,筆者らはTMEPAIファミリーに属しているC18ORF1もTGF-βによるEMTを抑制することをHuらと同じA549細胞を用いて明らかにしている7).HuらはTMEPAIの発現抑制系のみで結論づけており,大量発現系の結果を有していない点など,議論の余地がある.
3)TMEPAIによるPTENタンパク質発現抑制
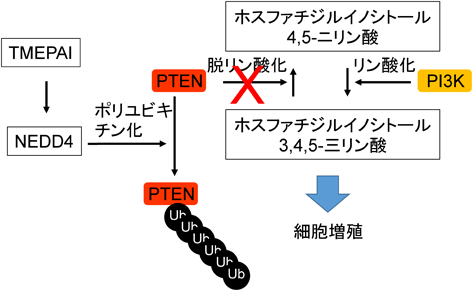
イノシトールリン脂質の脱リン酸化酵素であるPTENは,がん抑制遺伝子産物としてイノシトールに共有結合した3位のリン酸基を取り除く酵素活性を有する.エストロゲン受容体(ER),プロゲステロン受容体(PR)およびヒトEGF受容体関連2(human epidermal growth factor receptor type 2:HER2)のすべての発現が陰性であるトリプルネガティブの乳がん細胞でTMEPAIを大量発現させると,PTENの発現が抑制される.TMEPAIとPTENは直接結合することはできないが,NEDD4はTMEPAIとPTENに対する結合能を有しており,これらの三者複合体形成がPTENの分解につながると考えられている.三者複合体が形成されると,TGF-β刺激によりSmad非依存的シグナル系であるPI3K/Aktシグナル経路が活性化され,がん増殖ならびに転移が促進される45)(図9).
4)低酸素環境におけるTMEPAIの役割
腫瘍組織内部は,血管が十分に入り込めないために虚血状態になり,周辺の細胞は低酸素環境となる.低酸素環境で誘導されたTMEPAIは,HIF-1α(hypoxia-inducible factor-1α)依存的に活性化される転写反応を抑制している6).
1)TMEPAIの発現異常と疾患
大腸がん,乳がん,子宮がん,肺がん,腎細胞がんでは,TMEPAIの発現が亢進しているという報告がある1, 5, 46, 47).肺がん細胞を用いた実験では,TMEPAIの発現減少が細胞の増殖や転移能を減少させた48).また,乳がんのがん幹細胞維持にはTMEPAIの発現が必要であった49).前立腺がんではTMEPAIの発現が抑制されているという報告が多いが31, 35),6. 1)で述べたように,AR陽性の前立腺がんとAR陰性の前立腺がんでは,TMEPAI発現とがんの悪性化との関係が異なっている.前立腺がんにおけるTMEPAIの機能は他のがんに比較して解析が進んでいるが,相反する点も多い.多くのがんでTMEPAIの発現が亢進している理由としては,初期がんでTMEPAIの発現が上昇することでTGF-βの持つ細胞増殖抑制作用を阻害し,がん細胞自身がTGF-β存在下でも増殖できるような環境作りをしていることが考えられる.
2)C18ORF1発現と疾患
C18ORF1は,TMEPAIと異なり,がんとの関係についての報告はまったくない.しかしながら,C18ORF1遺伝子の転座や点変異と統合失調症の関連が取りざたされており,C18ORF1遺伝子が統合失調症感受性遺伝子の可能性も示唆されている50–52).TGF-βシグナルやARシグナルと統合失調症との関連については,よくわかっていない.
TMEPAIは,TGF-βシグナルのみならずARシグナルも抑制し,がん進展に関与することがわかってきている.また,HIF-1αシグナル系も抑制しており,さまざまなシグナルを抑制する多機能分子である.したがって,TMEPAIによるマルチシグナル抑制機構が明確になれば,TMEPAIを標的とした創薬開発にもつながると考えられる.C18ORF1遺伝子異常は統合失調症の発症に相関があるといわれているが,TGF-βシグナルが統合失調症に関与するとの報告はない.C18ORF1もマルチシグナル抑制分子である可能性がTMEPAIとの相同性からも類推できるので,統合失調症の原因となる細胞内シグナル伝達にC18ORF1が関与している可能性もある.筆者らは,すでにTMEPAIおよびC18ORF1ノックアウトマウスを有しているが,明確な表現型を見いだすことができていない.細胞は過度のTGF-βシグナルが伝達されないように,細胞外から核内までさまざまなステップで,TGF-βシグナル抑制分子が働いている.したがって,TMEPAIファミリー遺伝子を欠損しても,他のTGF-βシグナル抑制分子がその作用を補っていると思われる.同様にARシグナルなど他のシグナル系に関してもTMEPAIファミリー以外の分子が補完しているため,顕著な表現型を観察できていないのかもしれない.今後TMEPAIファミリーに関するさらなる機能解析が行われることで,そのマルチシグナル抑制機構が明確になっていくことを期待したい.
謝辞Acknowledgments
本研究で紹介した筆者の研究は,筑波大学大学院人間総合科学研究科実験病理学研究室所属時において加藤光保教授のご指導の下,多くの共同研究者とともに始めた研究であり,その後,昭和薬科大学薬学部生化学研究室で引き続き発展させてきたものです.特に現筑波大学大学院人間総合科学研究科実験病理学研究室助教 渡邊幸秀博士ならびに現昭和薬科大学薬学部生化学研究室助教 中野なおこ博士は,博士課程時より本研究に多大に貢献されており,この場を借りて両氏に深い感謝の意を表します.
引用文献References
1) Xu, L.L., Shanmugam, N., Segawa, T., Sesterhenn, I.A., McLeod, D.G., Moul, J.W., & Srivastava, S. (2000) Genomics, 66, 257–263.
2) Itoh, S., Thorikay, M., Kowanetz, M., Moustakas, A., Itoh, F., Heldin, C.H., & ten Dijke, P. (2003) J. Biol. Chem., 278, 3751–3761.
3) Watanabe, Y., Itoh, S., Goto, T., Ohnishi, E., Inamitsu, M., Itoh, F., Satoh, K., Wiercinska, E., Yang, W., Shi, L., Tanaka, A., Nakano, N., Mommaas, A.M., Shibuya, H., ten Dijke, P., & Kato, M. (2010) Mol. Cell, 37, 123–134.
4) Anazawa, Y., Arakawa, H., Nakagawa, H., & Nakamura, Y. (2004) Oncogene, 23, 7621–7627.
5) Giannini, G., Ambrosini, M.I., Di Marcotullio, L., Cerignoli, F., Zani, M., MacKay, A.R., Screpanti, I., Frati, L., & Gulino, A. (2003) Mol. Carcinog., 38, 188–200.
6) Koido, M., Sakurai, J., Tsukahara, S., Tani, Y., & Tomida, A. (2016) Biochem. Biophys. Res. Commun., 479, 615–621.
7) Nakano, N., Maeyama, K., Sakata, N., Itoh, F., Akatsu, R., Nakata, M., Katsu, Y., Ikeno, S., Togawa, Y., Vo Nguyen, T.T., Watanabe, Y., Kato, M., & Itoh, S. (2014) J. Biol. Chem., 289, 12680–12692.
8) Bai, X., Jing, L., Li, Y., Li, Y., Luo, S., Wang, S., Zhou, J., Liu, Z., & Diao, A. (2014) Cell. Signal., 26, 2030–2039.
9) Luo, S., Yang, M., Lv, D., Jing, L., Li, Y., Liu, Z., & Diao, A. (2016) Int. J. Biochem. Cell Biol., 76, 98–106.
10) Xu, L.L., Shi, Y., Petrovics, G., Sun, C., Makarem, M., Zhang, W., Sesterhenn, I.A., McLeod, D.G., Sun, L., Moul, J.W., & Srivastava, S. (2003) Cancer Res., 63, 4299–4304.
11) Li, H., Xu, L.L., Masuda, K., Raymundo, E., McLeod, D.G., Dobi, A., & Srivastava, S. (2008) J. Biol. Chem., 283, 28988–28995.
12) Li, H., Mohamed, A.A., Sharad, S., Umeda, E., Song, Y., Young, D., Petrovics, G., McLeod, D.G., Sesterhenn, I.A., Sreenath, T., Dobi, A., & Srivastava, S. (2015) Oncotarget, 6, 15137–15149.
13) Liu, R., Zhou, Z., Huang, J., & Chen, C. (2011) J. Pathol., 223, 683–694.
14) Derynck, R. & Miyazono, K. (2008) in The TGF-β Family (Derynck, R. & Miyazono, K. eds), pp. 29–43, Cold Spring Harbor Laboratory Press, New York.
15) Moses, H.L. & Roberts, A.B. (2008) in The TGF-β Family (Derynck, R. & Miyazono, K. eds.), pp. 1–28, Cold Spring Harbor Laboratory Press, New York.
16) Bierie, B. & Moses, H.L. (2006) Nat. Rev. Cancer, 6, 506–520.
17) 伊東 進(2012)がん増殖と悪性化の分子機構(宮澤,伊東編),pp. 27–41,化学同人.
18) Heldin, C.H. & Moustakas, A. (2016) Cold Spring Harb. Perspect. Biol., 8, a022053.
19) Hill, C.S. (2016) Cold Spring Harb. Perspect. Biol., 8, a022079.
20) Hata, A. & Chen, Y.G. (2016) Cold Spring Harb. Perspect. Biol., 8, a022061.
21) Shi, Y., Wang, Y.F., Jayaraman, L., Yang, H., Massagué, J., & Pavletich, N.P. (1998) Cell, 94, 585–594.
22) Xu, P., Liu, J., & Derynck, R. (2012) FEBS Lett., 586, 1871–1884.
23) Herhaus, L. & Sapkota, G.P. (2014) Cell. Signal., 26, 2186–2192.
24) Zhang, Y.E. (2009) Cell Res., 19, 128–139.
25) Derynck, R. & Zhang, Y.E. (2003) Nature, 425, 577–584.
26) Itoh, S., Landström, M., Hermansson, A., Itoh, F., Heldin, C.H., Heldin, N.E., & ten Dijke, P. (1998) J. Biol. Chem., 273, 29195–29201.
27) Nakano, N., Itoh, S., Watanabe, Y., Maeyama, K., Itoh, F., & Kato, M. (2010) J. Biol. Chem., 285, 38023–38033.
28) Reichling, T., Goss, K.H., Carson, D.J., Holdcraft, R.W., Ley-Ebert, C., Witte, D., Aronow, B.J., & Groden, J. (2005) Cancer Res., 65, 166–176.
29) Yoshikawa, T., Sanders, A.R., Esterling, L.E., & Detera-Wadleigh, S.D. (1998) Genomics, 47, 246–257.
30) Azami, S., Vo Nguyen, T.T., Watanabe, Y., & Kato, M. (2015) Biochem. Biophys. Res. Commun., 456, 580–585.
31) Fournier, P.G., Juarez, P., Jiang, G., Clines, G.A., Niewolna, M., Kim, H.S., Walton, H.W., Peng, X.H., Liu, Y., Mohammad, K.S., Wells, C.D., Chirgwin, J.M., & Guise, T.A. (2015) Cancer Cell, 27, 809–821.
32) Li, Y., Guo, A., Feng, Y., Zhang, Y., Wang, J., Jing, L., Yan, Y., Jing, L., Liu, Z., Ma, L., & Diao, A. (2016) Cell Prolif., 49, 710–719.
33) Nakano, N., Kato, M., & Itoh, S. (2016) J. Biochem., 159, 27–30.
34) Richter, E., Masuda, K., Cook, C., Ehrich, M., Tadese, A.Y., Li, H., Owusu, A., Srivastava, S., & Dobi, A. (2007) Epigenetics, 2, 100–109.
35) Sharad, S., Ravindranath, L., Haffner, M.C., Li, H., Yan, W., Sesterhenn, I.A., Chen, Y., Ali, A., Srinivasan, A., McLeod, D.G., Yegnasubramanian, S., Srivastava, S., Dobi, A., & Petrovics, G. (2014) Epigenetics, 9, 918–927.
36) Masuda, K., Werner, T., Maheshwari, S., Frisch, M., Oh, S., Petrovics, G., May, K., Srikantan, V., Srivastava, S., & Dobi, A. (2005) J. Mol. Biol., 353, 763–771.
37) Itoh, S. & Itoh, F. (2011) Growth Factors, 29, 163–173.
38) Itoh, S. & ten Dijke, P. (2007) Curr. Opin. Cell Biol., 19, 176–184.
39) Tsukazaki, T., Chiang, T.A., Davison, A.F., Attisano, L., & Wrana, J.L. (1998) Cell, 95, 779–791.
40) Itoh, F., Divecha, N., Brocks, L., Oomen, L., Janssen, H., Calafat, J., Itoh, S., & ten Dijke, P. (2002) Genes Cells, 7, 321–331.
41) Ding, Y. & Choi, M.E. (2014) Semin. Nephrol., 34, 62–71.
42) Jiang, Y., Woosley, A.N., Sivalingam, N., Natarajan, S., & Howe, P.H. (2016) Nat. Cell Biol., 18, 851–863.
43) Suzuki, H.I., Kiyono, K., & Miyazono, K. (2010) Autophagy, 6, 645–647.
44) Hu, Y., He, K., Wang, D., Yuan, X., Liu, Y., Ji, H., & Song, J. (2013) Carcinog., 34, 1764–1772.
45) Singha, P.K., Yeh, I.T., Venkatachalam, M.A., & Saikumar, P. (2010) Cancer Res., 70, 6377–6383.
46) Brunschwig, E.B., Wilson, K., Mack, D., Dawson, D., Lawrence, E., Willson, J.K., Lu, S., Nosrati, A., Rerko, R.M., Swinler, S., Beard, L., Lutterbaugh, J.D., Willis, J., Platzer, P., & Makowaitz, S. (2003) Cancer Res., 63, 1568–1575.
47) Rae, F.K., Hooper, J.D., Nicol, D.L., & Clements, J.A. (2001) Mol. Carcinog., 32, 44–53.
48) Vo Nguyen, T.T., Watanabe, Y., Shiba, A., Noguchi, M., Itoh, S., & Kato, M. (2014) Cancer Sci., 105, 334–341.
49) Nie, Z., Wang, C., Zhou, Z., Chen, C., Liu, R., & Wang, D. (2016) Acta Biochim. Biophys. Sin. (Shanghai), 48, 194–201.
50) Kikuchi, M., Yamada, K., Toyota, T., Itokawa, M., Hattori, E., Yoshitsugu, K., Shimizu, H., & Yoshikawa, T. (2003) Mol. Psychiatry, 8, 467–469.
51) Kikuchi, M., Yamada, K., Toyota, T., & Yoshikawa, T. (2003) J. Med. Dent. Sci., 50, 225–229.
52) Meerabux, J.M., Ohba, H., Iwayama, Y., Maekawa, M., Detera-Wadleigh, S.D., DeLisi, L.E., & Yoshikawa, T. (2009) J. Hum. Genet., 54, 386–391.
著者紹介Author Profile
 伊東 進(いとう すすむ)
伊東 進(いとう すすむ)昭和薬科大学薬学部生化学研究室教授.薬学博士.
略歴1962年石川県金沢市生まれ,85年大阪大学薬学部薬学科卒業,90年同大学院薬学研究科博士後期課程修了(大阪大学細胞工学センター谷口維紹教授の下で研究に従事),90年薬学博士,同年北海道大学薬学部代謝分析学講座助手(鎌滝哲也教授),94年大阪大学薬学部微生物薬品化学講座助手(三村務教授),96年Sweden Ludwigがん研究所研究員(Carl-Henrik Heldin教授),99年オランダ王国がん研究所研究員(Peter ten Dijke博士),2002年筑波大学大学院人間総合科学研究科実験病理学講座准教授(加藤光保教授),10年より現職.
研究テーマと抱負TGF-βシグナル伝達とがん進展に関する研究.
ウェブサイトhttp://www.shoyaku.ac.jp/research/laboratory/seika