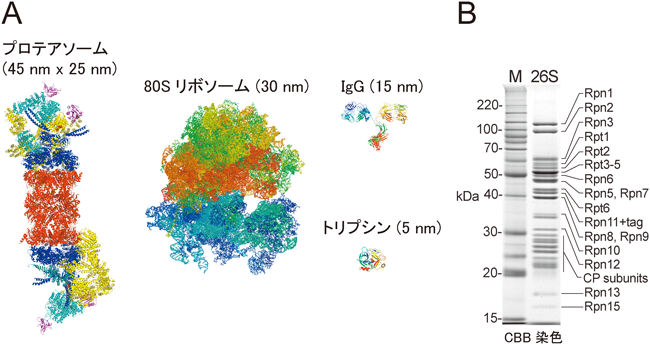
プロテアソームはプロテアーゼ活性を持つ触媒ユニットCP(core particle,別称20Sプロテアソーム)とユビキチン化タンパク質の分解に必須の制御ユニットRP(regulatory particle,別称PA700)が会合した構造を持ち,この活性化型フォームを26Sプロテアソームまたは単にプロテアソームと呼ぶ(余談だが,この26Sの“S”は沈降係数のSであり,CPの片側にRPが会合したRP1-CPは26 S,CPの両端にRPが会合したRP2-CPは30 Sであるため,正確には両者を分けて呼ぶべきであるが,機能に差異があるか不明であり,今のところ慣例的にどちらも26Sプロテアソームと呼ばれている)1,2,4).プロテアソームは非常に不安定な酵素であり,生化学的解析および構造解析が大きく立ち遅れていた.実際,最後の構成サブユニットRpn15/Sem1を筆者らが同定したのは2004年である5).そのころ,エピトープタグ融合によるプロテアソームのアフィニティー精製法が開発され,個々のサブユニットやPIPsの機能解析が大きく進展した6–8).プロテアソームサブユニットのストイキオメトリ(化学量論)について,Robinsonらと共同で30SプロテアソームをNative質量分析計により解析したところ,その質量は2,587,352 Daと決定された9).この値は33種類の構成サブユニットを2分子ずつ含んだ理論値とほぼ一致したため,プロテアソームの全構成サブユニットが確定した(図1B).
CPは,出芽酵母およびウシ肝臓由来のものについて結晶構造解析がなされ,触媒活性部位の解明のみならずプロテアソーム阻害剤の開発に貢献してきた10,11).その後,当然の課題として筆者らを含む多くの研究者がプロテアソーム全体の結晶化に挑戦したが残念ながらことごとく失敗に終わった.これはプロテアソームが複雑な形状を持つこと,26Sと30 Sを分離することが難しいこと,さらにはPIPsによる試料の不均一性の問題があるためである.このような高難度タンパク質複合体では,結晶構造解析に替わる方法としてクライオ電子顕微鏡による単粒子解析が有効である.筆者らは出芽酵母プロテアソームのアフィニティー精製法を開発し,マックスプランク研究所のBaumeisterらと共同でクライオ電子顕微鏡による単粒子解析を行い,プロテアソームのユビキチン受容体Rpn10とRpn13の分子内の位置を決定した12).現在では,プロテアソームのクライオ電子顕微鏡の密度マップ(7~10 Å)をもとに原子構造モデルが作製されている(図2).その後も,BaumeisterらのグループとUCバークリー校のMartinらのグループが激しく競争しつつも精力的にプロテアソームの構造解析を推進しており,驚異的なスピードで次々と新知見が得られている13,14).
1)プロテアソームの原子構造モデル
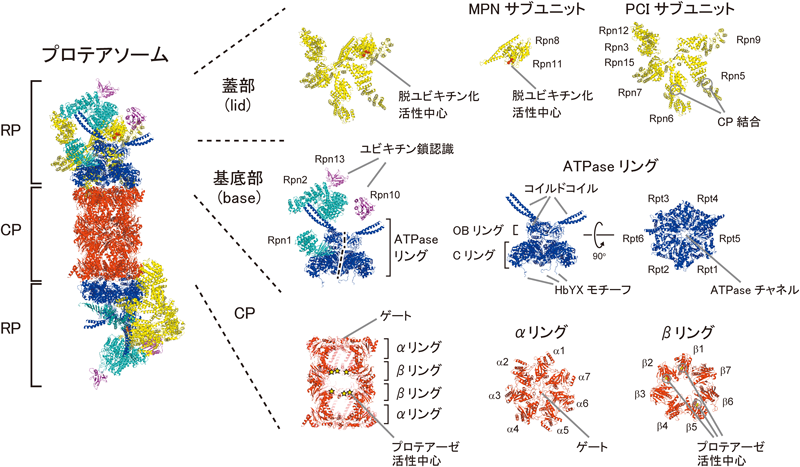
プロテアソームの高分解能電子顕微鏡マップに各サブユニットの結晶構造モデルを当てはめることで全体の原子構造モデルが作製されている(図2)15).一見してプロテアソームが非常に精緻かつむだのない美しい構造を持つことがわかる.CPはそれぞれ七つのサブユニットからなるαリングとβリングがαββαの順に連なった円筒構造を持ち,計14種類28個のサブユニットからなる複合体である.プロテアソームはThr型プロテアーゼに分類され,β1,β2,β5が持つ活性中心は分子内部に隔離されている.基質の入り口である狭い孔はαサブユニットのN末端テールにより形成されるゲートでふさがれており,CP単独では基本的に不活性であるためself-compartmentalizing proteaseと表される16).RPは六つのATPaseサブユニットと13個のnon-ATPaseサブユニットの計19種類の構成サブユニットを持ち,ユビキチン化タンパク質の分解において,ユビキチン鎖の認識,基質タンパク質のアンフォールディング,ユビキチン鎖の除去,解きほぐした基質タンパク質のCP内腔への送り込みといった機能を持つ.RPのサブユニット名は種によって異なり若干混乱するが,解析が先行している出芽酵母の統一名称を用いるのがまだ一般的であり,本稿ではATPaseサブユニットはRpt(regulatory particle triple-A ATPase),non-ATPaseサブユニットはRpn(regulatory particle non-ATPase)で統一している17).なお,Rpn4とRpn14は当初プロテアソームの構成サブユニットとして同定されたが,Rpn4はプロテアソーム遺伝子発現を制御する転写因子,Rpn14はRP専用シャペロンであることが後に明らかとなった18,19).
RPは生化学的にATPaseサブユニットを含む基底部(base)と脱ユビキチン化酵素サブユニットRpn11を含む蓋部(lid)の二つのサブ複合体に分離できる.基底部は六つのATPaseサブユニット(Rpt1~6)と四つのnon-ATPaseサブユニット(Rpn1, Rpn2, Rpn10, Rpn13)から構成される.このうちATPaseサブユニットは二つずつのペア(Rpt1–Rpt2, Rpt6–Rpt3, Rpt5–Rpt5)の三量体としてATPaseリングを形成しており,CPのαリングの上に鎮座している20).各RptはN末端領域に長く伸びたコイルドコイル,中央にOBドメイン,C末端領域にATPaseドメインを持ち,ATPaseリングはOBリングとATPaseドメインからなるCリングの2層構造を持つ.現在,基質非存在下のプロテアソーム(待機状態)および基質存在下で活性化状態にあるプロテアソームの高次構造モデルが得られている15,21–23).待機状態のプロテアソーム中ではCリングはRpt3を頂点,Rpt2を底としたらせん階段構造をとっており,ATPaseチャネルの軸はCPチャネルの軸から約10度傾いている.一方,活性化状態のプロテアソーム中では各RptサブユニットのATPaseドメインが平面に並び,Cリングが直立することで基質タンパク質の通り路となる各チャネルの軸が一直線につながる.基質と結合するとプロテアソームのATPase活性は上昇するが,実際,ATPγS(ATPの非加水分解性アナログ)存在下の構造は活性化構造とよく似ていた24).このATP配位によるCリングの構造変化はRpt6–Rpt3ペアのコイルドコイル領域とRpn2を介してRP全体の構造変化を引き起こす.また,Rpt2,Rpt3,Rpt5のC末端はHbYX(疎水性アミノ酸-Tyr-任意のアミノ酸)配列を持ち,このモチーフがCPのαリングの窪みに突き刺さることで基底部-CP複合体を安定化させるとともにアロステリックにCPのゲートを開かせる機能を持つ25–27).基底部は二つの大きなnon-ATPaseサブユニットRpn1とRpn2を持ち,Rpn1は脱ユビキチン化酵素Ubp6(ヒトではUSP14)やユビキチン化基質シャトリング分子Rad23などのUBL(ubiquitin-like)ドメインを持つPIPsの受容体として機能し7,28,29),Rpn2はユビキチン受容体サブユニットRpn13の受容体として機能する30–34).もう一つのユビキチン受容体サブユニットRpn10はRP形成の最終段階で取り込まれること,蓋部および基底部の双方と相互作用することから厳密ではないのだが基底部の構成サブユニットとして定着している1,4).
蓋部(lid)は必須の脱ユビキチン化酵素サブユニットRpn11を含む九つのnon-ATPaseサブユニットからなる複合体であり,高塩濃度存在下でプロテアソームから遊離するサブ複合体として報告された35,36).類似した複合体としてeIF3やCOP9/CSN/シグナロソームが知られており,蓋部はPCI(proteasome-CSN-eIF3)ドメインを持つサブユニット(Rpn12–Rpn3–Rpn7–Rpn6–Rpn5–Rpn9)とMPN(Mpr1, Pad1 N-terminal)ドメインを持つサブユニット(Rpn8–Rpn11),および最小サブユニットのRpn15(Sem1/Dss1)に分類される.当初,蓋部はATPaseリングの上部に位置し蓋のような構造を持つ(プロテアソームのイラストではよくゴミ箱の蓋として描かれる)と想定されていたためそのように命名されたが,構造解析の結果,実際は馬蹄鉄型の構造を持ち,基底部を側面から覆うように配置することが明らかとなった15,21,35).蓋部はATPaseチャネルの真上にRpn11を配置させるほか,Rpn5とRpn6がCPと直接相互作用することによりプロテアソームの構造を安定化させている.Rpn15/Sem1(ヒトではDSS1/SHFM1)は表面が酸性電荷を帯びた89アミノ酸の小さな不思議なサブユニットである5,37,38).Rpn15はTREX-2複合体やTHP3-CSN12複合体,BRCA2複合体などプロテアソーム以外のさまざまな複合体に含まれており,特にBRCA2複合体中ではDNA二本鎖切断の相同組換え修復に関与している39–41).いずれも核酸結合能を持つ複合体であるため,Rpn15は核酸ミミックとして何らかの機能を果たしていると考えられる42).また,分裂酵母のRpn15がユビキチン結合能を持つことが最近報告されたが,Rpn15はRpn3とRpn7サブユニット間の溝に埋もれていること,RPのユビキチン化基質プロセシング部位の裏側に位置することから,プロテアソーム内でユビキチン受容体として機能しているかは不明である43–45).
2)プロテアソームによる基質分解機構
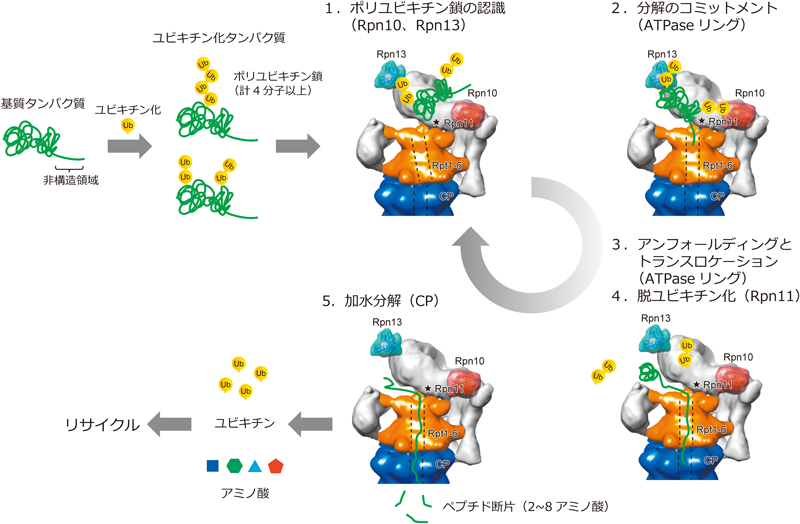
プロテアソーム分解はRPによるユビキチン鎖の認識,基質タンパク質の解きほぐしとCP内への送り込み,ユビキチン鎖の除去,CP内での加水分解に分けることができ,それぞれ生化学的に解析されてきたが,それぞれの反応がどのように統合されているかは不明であった.しかし最近,プロテアソームの高次構造が明らかとなり,ATPaseリングの構造変化に駆動されて各反応が協調して起こることがわかってきた.ここではまずプロテアソームの分解シグナルについて最新の知見を紹介した後,プロテアソームの作動機構をユビキチン鎖認識,基質タンパク質の認識と解きほぐし,脱ユビキチン化そして基質タンパク質の分解の五つにわけて紹介する.次いで,プロテアソーム結合タンパク質PIPsによるプロテアソーム分解の調節機構を紹介する.
a. プロテアソームの分解シグナル
四つ以上のユビキチンが連結したLys48リンク型のポリユビキチン鎖がプロテアソームの分解シグナルとなることが2000年に報告されて以来,セントラルドグマとして長い間信じられてきたが,最近では少し状況が変わってきている46,47).まず,Ciechanoverらは150アミノ酸以下の小さなタンパク質は,モノユビキチン化あるいはマルチプルモノユビキチン化(複数箇所のモノユビキチン化)で十分プロテアソームにより分解されることを報告した48).これは網状赤血球の無細胞系あるいは試験管内の実験ではあるが,種々のモデル基質,CDKの小サブユニットCks2(79アミノ酸),αシヌクレイン(140アミノ酸),酵母のHug1(68アミノ酸)などはモノユビキチン化で確かに分解される.また,NFκB p50サブユニットの前駆体p105はマルチプルモノユビキチン化でプロテアソームにより限定分解を受けることが知られている49).一方,M期進行に関わるユビキチンリガーゼAPC/CとサイクリンBを用いた再構築系では,短いユビキチン鎖がサイクリンBの複数のリシン残基に付加されるが,ごく最近Kirschnerらは,ユビキチン修飾のトポロジーに依らず計4分子以上のユビキチンが付加することがプロテアソーム分解に重要であることを報告した50–52).少し詳しく説明すると,この論文ではサイクリンBおよびユビキチンを異なる蛍光色素で標識し,ユビキチン化およびプロテアソーム分解を蛍光一分子イメージングにより定量的に解析している.サイクリンBに付加するユビキチン分子数は四つに固定して,テトラユビキチン鎖1本,ジユビキチン鎖2本,ジユビキチン鎖1本およびモノユビキチン化2か所の効果を比較したところ,プロテアソームによるサイクリンBの捕捉・分解速度はほぼ同等であった.一方,マルチプルモノユビキチン化4か所の場合はプロテアソームによる捕捉時間は短鎖と同じだが分解されない.つまり,プロテアソームによるターゲティングにはユビキチン密度が重要であり,その後のステップにユビキチン鎖が必須である.これらの報告は従来のモデルを大きく覆すものだが,実は細胞内におけるプロテアソーム基質のユビキチン修飾の構造(鎖のタイプや鎖長)についてはまだまだ知見が乏しいため,あながち間違っているとはいえない.実際,質量分析計を用いた網羅的な解析より,ユビキチン化基質の約60%はユビキチン化部位が複数箇所あること,またプロテアソーム阻害時でも全ユビキチンの25%程度しかポリユビキチン鎖形成に使われていないことが報告されている53,54).つまり,ほとんどのユビキチン化基質はマルチプルモノユビキチン化と少量のポリユビキチン鎖が付加した不均一な修飾を受けているようである.また,Lys48リンクのポリユビキチン鎖がもっぱらプロテアソーム分解に関与するとされているが,試験管内ではLys63リンクのポリユビキチン鎖,Lys11リンクとLys48リンクが混ざった分岐鎖は十分プロテアソーム分解を促進するため,細胞内でプロテアソーム基質がどのようなユビキチン修飾を受けるのか精査する必要に迫られている47,55–58).
また,基質タンパク質自体の構造的安定性がプロテアソーム分解の重要なパラメーターの一つであるらしい.ユビキチン化されればどんなタンパク質でもプロテアソームにより分解されるわけではなく,基質タンパク質自身が20~30アミノ酸の長さのふらふらした非構造(unstructured)領域を持つことが重要であることが示されている59,60).この2デグロンモデルはプロテアソーム分解のコミットメントステップ(後述)をうまく説明できるため広く受け入れられている.それではこの非構造領域には何か分解に適した特徴的な配列が存在するのだろうか? ごく最近の報告によると,意外なことにアミノ酸組成の複雑さのみが分解速度と相関した.つまり,非構造領域のもつ電荷や疎水性度,ヘリックスの割合,体積などはあまり関係なく,異なるアミノ酸が適度に分布していることが重要であるらしい61).
b. プロテアソームの作動機構
ⅰ)プロテアソームによるユビキチン鎖の捕捉ユビキチン受容体サブユニットRpn10とRpn13はATPaseチャネル近傍に位置すると予想されていたが,筆者らはBaumeisterらとの共同研究により,RPの端に互いに離れて存在することを明らかにした(図2)12,62).この分子内配置はユビキチン化基質を捕捉するのに有利と考えられる.得られた構造モデルをもとに機能サブユニット間の距離をみてみると,Rpn10のユビキチン結合モチーフUIM(ubiquitin-interacting motif)とRpn13のユビキチン結合ドメインPru(pleckstrin-like receptor of ubiquitin)間の距離は約90 Å,Rpn11の活性中心までの距離はそれぞれ約90 Å,70 Åであった.UIMおよびPruドメインはユビキチンのIle44を含む疎水性パッチを認識しユビキチン単量体と1 : 1で相互作用するため,単独ではユビキチン鎖タイプの選択性はあまりみられない33,34).Lys48リンクのテトラユビキチンの場合最短で約70 Åと計算されるため,Rpn10でもRpn13でもそれぞれ基質タンパク質をATPaseチャネルに提示することが可能である.一方,Rpn10とRpn13は遺伝学的にも生化学的にも協調的に機能することが示唆されているため,Rpn10とRpn13が同時に1本のポリユビキチン鎖を捕捉する可能性もある12).
では前述のCiechanoverらのモノユビキチン化のみでプロテアソーム分解が促進されるというモデルはプロテアソームの構造から説明できるのだろうか? ユビキチン単量体は約25 Åであり,Rpn10 UIM,Rpn13 PruとATPaseチャネルの入り口までの距離はそれぞれ約90 Å,100 Åであるため,構造を持たない基質タンパク質なら,モノユビキチン化のみでもATPaseチャネルまで届きそうである.実際,Rpn10を欠損させたプロテアソームではモノユビキチン化基質の分解が遅延する.一方,Kirschnerらのモデルでは基質に複数の短いポリユビキチン鎖が存在するため,プロテアソームとユビキチン化基質はアビディティー(avidity)で相互作用することになる(図3).プロテアソームのユビキチン結合価は2価(Rpn10とRpn13)であるため,ユビキチン鎖が複数ある方が捕捉される確率が上がる.さらに短鎖の場合それぞれが弱い相互作用なので,基質タンパク質はプロテアソーム上を比較的自由に動けると考えられ,非構造領域がATPaseチャネルに入り込む確率が高くなるだろう.ユビキチン鎖がRpn10とRpn13と強固に結合すると基質タンパク質の分解の際に阻害的に働くことが想定されるが,短鎖ならその問題も解消されるため,筆者には魅力的なモデルに思える.
なお,出芽酵母の遺伝学的な解析では,Rpn10,Rpn13,さらにはRad23などのシャトリング分子をすべて同時に破壊しても致死とはならないため,プロテアソーム内にはまだユビキチン結合部位が存在すると考えられている.
ⅱ)プロテアソームによる分解のコミットメントATPaseチャネルの入り口から60~70 Åの位置に各Rptは基質と直接相互作用するAr-φループを持つ.先に述べたようにプロテアソームで効率よく分解されるためには基質タンパク質が20アミノ酸以上(70 Å以上)の非構造領域を持つことが重要であり,ちょうど距離的に一致する60).基質タンパク質の非構造領域がAr-φループに捕捉されることで,プロテアソームは基質タンパク質を分解することを決定し,ATPase活性が上昇する.この分解のコミットメントステップは概念としては以前より存在したが,生化学的に確認されたのはつい最近のことである63,64).Pethらはプロテアソームとポリユビキチン化タンパク質の相互作用をほぼリアルタイムで高感度に検出する系を開発した.その結果,プロテアソームの基質認識は2段階で起こっており,最初にユビキチン受容体に依存したユビキチン鎖の認識が,次いでATP依存的かつ高塩濃度耐性の強い相互作用が観察された.後者の結合は非構造領域を持つタンパク質に特異的にみられたことから,ATPaseサブユニットが基質タンパク質の非構造領域を捕捉していることが証明された64).
ⅲ)アンフォールディングとトランスロケーションCPチャネルは約13 Åと狭いため,基質タンパク質はATPaseチャネルを通過する過程で立体構造が破壊されなければならない10,16).基質をアンフォールディング中の高次構造は得られておらず詳細はまだ不明だがおおむね次のように考えられている22,24,27).分解のコミットメントにより,プロテアソームは活性化型に構造変化し基質の通り路がつながる(トランスロケーション許容構造と呼ばれる).このとき,プロテアソームはATPが複数のRptサブユニットに配位した遷移状態であり,次いでATPの加水分解が順番にあるいはランダムに起こり各ATPaseドメインがピストンのように上下に動く65).この際,基質をつかんだAr-φループも駆動するが,強固な構造を持つOBリングが分子枷となり基質タンパク質の高次構造が破壊されていく66,67).基質タンパク質はATPの加水分解サイクルにより解きほぐされると同時にCP内部に送り込まれる.なお,六つのRptサブユニットの機能は等価ではなく,基質のアンフォールディング,トランスロケーション,CP活性化にはそれぞれ異なる組み合わせのRptが協調的に働くようである27).Martinらは大腸菌共発現系による基底部の再構築に成功し,さまざまな変異を導入することで各Rptサブユニットの機能を反応速度論的に徹底的に解析した27).その結果,基質のアンフォールディングの開始にはRpt3,Rpt4,Rpt6のATP加水分解が必要であり,基質のトランスロケーションにはRpt1が最も寄与していた.また,CPのゲートを完全に開くにはRpt2,Rpt3,Rpt5のHbYXモチーフが同時にCPに刺さることが必要らしい.
ⅳ)脱ユビキチン化脱ユビキチン化酵素サブユニットRpn11の活性中心はATPaseチャネルの真上に位置し,ユビキチン化基質のユビキチン鎖を根元から削ぎ落とすようにして脱ユビキチン化する22,23,68).このRpn11による脱ユビキチン化は完全にATP依存的であり,基質タンパク質のトランスロケーションと協調して起こる69).待機状態ではRpn11の活性中心はRpt4-Rpt5のコイルドコイルの近くに位置し活性が阻害されている.しかし,基質タンパク質のアンフォールディングに伴いプロテアソームが活性化型構造になると,ATPaseリングの構造変化がRpt3–Rpt6のコイルドコイルを介してRpn2に伝わり,蓋部全体が約25度回転する.これに伴いRpn11の活性中心が約18 ÅスライドしATPaseチャネルの真上10 Åの位置に移動する.この状態では基質タンパク質はユビキチンが除去されない限りATPaseチャネルに入れない.つまり,Rpn11による脱ユビキチン化はユビキチン化基質分解の律速段階であり,Rpn11はATPaseチャネルの門番として機能することになる.最近,MPNサブユニットRpn11とRpn8の共結晶構造が解かれ,脱ユビキチン化反応の詳細が明らかとなった68,70).Rpn11はJAMMファミリーに属するメタロプロテアーゼであり,同ファミリーの脱ユビキチン化酵素と比較してユビキチンとの結合は非常に弱く,それゆえに活性中心近傍に提示されるユビキチンを非選択的に捕まえ切断できることが示された.なお,切り出されたユビキチン鎖は細胞内の脱ユビキチン化酵素により単量体にまで切断され再利用される71).
ⅴ)CPによる加水分解ATPaseチャネルを通過する過程で解きほぐされた基質タンパク質はCPゲートを通過した後,まずantechamberと呼ばれる前室に送り込まれる.antechamberは基質タンパク質がリフォールディングして再び構造を持たないように変性状態を保つ機能を持つ72).そして基質タンパク質はβ1,β2,β5の活性中心が存在するcatalytic chamberに送り込まれ,2~8アミノ酸のオリゴペプチドに加水分解される.活性中心に基質タンパク質が接触しているとRP–CP間の相互作用が安定化されることが示唆されており,これは基質タンパク質の不完全な分解を防ぐためと考えられている73).プロテアソームから排出されたペプチドはTPPIIやアミノぺプチダーゼによりアミノ酸まで分解され再びタンパク質の生合成に用いられる74).
3)プロテアソーム結合タンパク質によるプロテアソーム分解の制御
プロテアソームの構成サブユニットではないが,プロテアソームと一過的に結合するタンパク質群PIPsが多数存在する.ここではPIPsのうち,プロテアソーム分解を制御する主要な分子を紹介する.
ⅰ)ユビキチン化基質シャトリング分子
細胞内でポリユビキチン化基質はプロテアソームに直接認識される場合と,Rad23(ヒトではRAD23A,RAD23B)やDsk2(UBQLN1, 2, 4)のようなシャトリング分子により捕捉されプロテアソームに運搬される場合がある1).これらのシャトリング分子はUBL(ubiquitin-like)ドメインを介してRpn1(ヒトではRpn10とRpn13も関与)と結合する一方,UBA(ubiquitin-associated)ドメインを介してポリユビキチン鎖と結合する.特にRad23はユビキチン選択的シャペロンのCdc48(VCP/p97)の下流で機能するとされており,広範な基質を細胞中からプロテアソームに運搬していると想定されている75).これらのシャトリング分子の基質選択性や使い分けは未だ良く分かっておらず今後の解析が待たれている.
また,高等真核生物ではUBL–UBAタンパク質以外にも,ヒ素で誘導されるAIRAPL/ZFAND2B,筋萎縮に関与するZNF216/ZFAND5,チオレドキシン様タンパク質TXNL1がシャトリング分子として報告されている76–78).
ⅱ)脱ユビキチン化酵素
プロテアソームの主要なPIPsとして脱ユビキチン化酵素Ubp6(ヒトではUSP14)とUCH37/UCHL5が知られている1).Ubp6とUCH37はプロテアソームと結合すると活性化し,プロテアソーム上でユビキチン化基質のポリユビキチン鎖をトリミングする7,32).このユビキチン鎖のトリミングは基質タンパク質のユビキチン化の状態により基質の分解を負にも正にも制御する.ユビキチン鎖のトリミングが基質分解のコミットメントより先に起こると基質タンパク質は分解から免れる「ユビキチンタイマーモデル」では負に働くだろうし,長すぎるユビキチン鎖や分岐型ユビキチン鎖が付加した基質に対してはプロテアソームがハンドリングしやすい長さにユビキチン鎖をトリミングしたほうが基質の分解を促進させるだろう.ごく最近,Ubp6-プロテアソーム複合体の高次構造が報告され,Ubp6がユビキチンセンサーとしてプロテアソームの構造を直接制御していることが明らかとなった79,80).Ubp6はN末端のUBLドメインを介してRpn1と結合するが,C末端領域のUSP(ubiquitin specific protease)ドメインもユビキチンが結合するとプロテアソームのATPaseリングと相互作用し活性化型構造(ATP配位型)を安定化させる.興味深いことに,このときRpn11活性および基質のトランスロケーションは阻害されており,Ubp6活性が優位となっている.つまり,Ubp6にユビキチンが接触している間はUbp6がプロテアソーム分解を支配していることになる.ユビキチン修飾の構造に依存するが,いずれにしてもUbp6は分解のタイムキーパーとして機能することが明確となった.また,まったく別の可能性として,活性化状態のプロテアソームには他のユビキチン化基質が結合できないことから,Rpn11により切り出されたユビキチン鎖がUbp6にトラップされることで,しばらくプロテアソームの活性化状態を維持し,次のユビキチン化基質が結合するタイミングを調節しているのかもしれない80).後述のとおり,ヒトホモログUSP14の阻害剤IU1は易凝集性タンパク質の分解を促進させることが報告されており,Ubp6/USP14は非常に興味深いPIPsの一つである81).
UCH37/UCHL5はもう一つの脱ユビキチン化酵素で,Rpn13を介してプロテアソームと結合する30–32).UCH37は分裂酵母や高等真核生物には存在するが出芽酵母には存在しない.同様に,出芽酵母のRpn13はUCH37結合ドメインを欠いた構造を持つ.UCH37はプロテアソームと結合すると活性が亢進するが,特に短いユビキチン鎖をdistal側から切断することが知られている82).UCH37はプロテアソーム以外にもクロマチンリモデリングに関与するINO80複合体にも存在する.INO80複合体中では脱ユビキチン化活性が完全に抑制されているが,プロテアソームを共存させると脱ユビキチン化活性が亢進するため,プロテアソームはINO80複合体とともに転写やDNA修復に関与することが示唆されている83–85).
ⅲ)ユビキチンリガーゼ
プロテアソームと相互作用するユビキチンリガーゼがいくつか同定されている.出芽酵母のHul5(ヒトではUBE3C)はユビキチン鎖の伸長活性(E4活性)を持ちUbp6と拮抗することから,プロテアソーム上で基質のユビキチン化を亢進させ分解を促進させるというモデルが提唱されている86).さらにヒトではUBE3A/E6APやHERC2,HUWE1,UBR4,RNF181などの巨大なユビキチンリガーゼがプロテアソームと相互作用することが知られている87–89).これらのユビキチンリガーゼがプロテアソームと直接相互作用する意義はよくわかっていないが,UBE3AやUBE3Cはプロテアソーム(特にRpn10,Rpn13,Rpt5,UCH37)をユビキチン化することで,プロテアソームのユビキチン鎖結合能や脱ユビキチン化能を低下させる,つまりプロテアソームの分解活性を負に制御することが報告されている88,89).このプロテアソームのユビキチン化は酸化ストレスや熱ストレスあるいは飢餓状況下で一過的に起こる可逆的な現象であり,プロテアソーム活性を制御する新しい機構であると考えられている.
プロテアソーム分解はタンパク質恒常性の維持に必須の役割を果たすが,ストレス応答や細胞分化に伴いプロテアソーム分解のキャパシティーがダイナミックに変動することがわかってきた.分解キャパシティーを拡大縮小するためには,先に述べたようにPIPsによるプロテアソーム活性の制御があるが,細胞は主にプロテアソームの量を調節する戦略をとるようである.ここではまず,プロテアソーム遺伝子の発現調節機構とプロテアソームの形成機構について紹介し,次いでプロテアソームの安定化因子,プロテアソーム自身の代謝,細胞分化や加齢に伴うプロテアソームレベルの変動について紹介する.
1)プロテアソーム遺伝子の発現制御機構
プロテアソームは前述のとおり33種類以上のサブユニットから形成するため,個々のサブユニット遺伝子が一斉に転写翻訳される必要がある.プロテアソームサブユニット遺伝子の発現を誘導する転写因子として出芽酵母ではRpn4が同定されている18,90).Rpn4はC2H2ジンクフィンガー型の転写因子でありプロテアソーム関連遺伝子のプロモーター上にあるPACE(proteasome-associated control element)配列を認識し転写を誘導する91).RPN4の破壊株ではプロテアソームの量が約半分に低下する.Rpn4の発現は熱ストレスや酸化ストレス,アミノ酸アナログなどのタンパク質毒性ストレス,DNA損傷によって誘導され,プロテアソームの量を増加させる.興味深いことに,Rpn4自身がプロテアソームの基質であり,細胞内のプロテアソーム量が増加するとRpn4の量が減少する.つまり,プロテアソーム活性とRpn4量が逆相関する負のフィードバックループが存在し,必要に応じて適切な量のプロテアソームが発現する92).
高等真核生物ではRpn4は存在しないが機能的なホモログとしてNrf1/NFE2L1が報告された93–97).Nrf1は酸化ストレス応答に関わるロイシンジッパー型の転写因子であり小胞体膜上にアンカーされている.通常,Nrf1は小胞体関連分解(ER associated degradation: ERAD)経路でユビキチン化されプロテアソームにより恒常的に完全分解されている.しかし,プロテアソーム機能が低下するとNrf1はプロテアソームにより限定分解を受けて小胞体から遊離し活性化する.活性化したNrf1は核へと移行しプロテアソーム遺伝子の発現を誘導する.プロテアソーム遺伝子はNrfファミリー(Nrf1, Nrf2, Nrf3)が認識する共通の配列AREs(antioxidant response elements)を持つが,Nrf1のみがプロテアソーム遺伝子の発現を誘導する.酵母のRpn4とは異なり,Nrf1ノックアウト細胞ではプロテアソーム量は減少しないため,基底レベルでのプロテアソーム発現制御は他の転写因子が担っていると考えられている93,94).一方,高等真核生物では,後述のようにFOXO4やマイクロRNAによる単一サブユニットの発現制御によりプロテアソーム量が調節されることもわかってきた98,99).
2)プロテアソームの分子集合
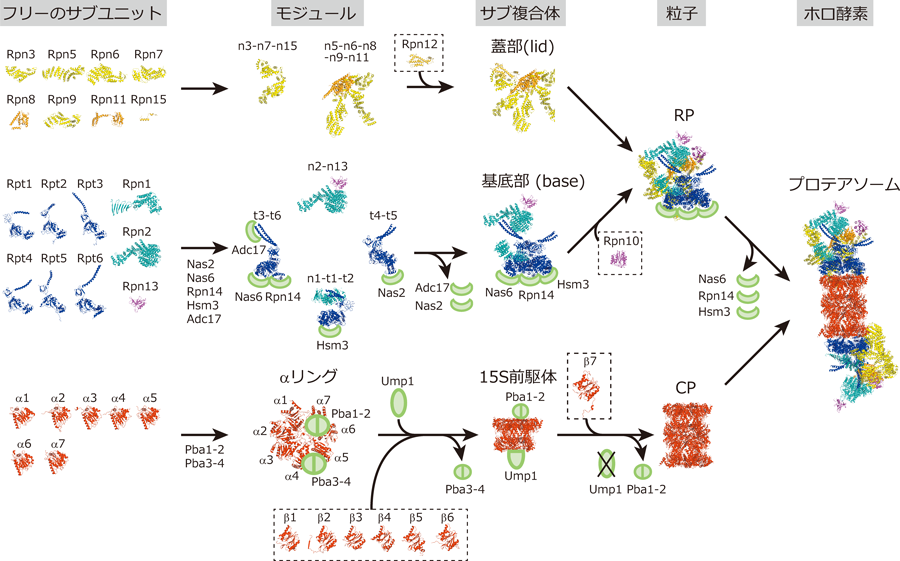
細胞内はタンパク質が充満した非常に込み入った環境であり,プロテアソームのような超分子複合体が正確に形作られるのは奇跡のように思える.プロテアソームの分子集合は隣接する二つないし三つのサブユニットのペアによるサブユニット間の相互作用から始まるが,ここで間違った組み合わせが生じると,完成品には取り込まれないdead-end productや,間違った組成と構造を持つ有害なプロテアソームを生じてしまい細胞にとって致命的である.2005年,村田らによりPAC1–PAC2(proteasome assembling chaperone 1–2)が発見され,細胞内にはプロテアソーム形成のための専用のシャペロンが存在し,プロテアソームの分子集合を支援していることが明らかとなった100).その後,プロテアソームの分子集合研究は世界中で大きく進展し,これまで,CPの専用シャペロンとしてPba1–Pba2(ヒトではPAC1/PSMG1-PAC2/PSMG2),Pba3–Pba4(PAC3/PSMG3–PAC4/PSMG4),Ump1(POMP)の五つの分子が,RP専用シャペロンとしてNas2(p27/PSMD9),Nas6(p28/gankyrin/PSMD10),Rpn14(PAAF1),Hsm3(S5b/PSMD5),Adc17(ヒトには相同遺伝子なし)の五つの分子が同定されている4,101–103).ここでは,CPとRPの形成機構をそれぞれ紹介し,さらに分子シャペロンによる制御について紹介する.
ⅰ)CPの形成機構
CPの形成はαリングの形成から始まる.解析が先行している酵母では,Pba3–Pba4ヘテロ二量体が七つのαサブユニットのうちα5と特異的に結合し,さらにα4を引き寄せることでα4–α5複合体を形成させる4,104,105).このα4–α5を核として残りのαサブユニットが順次結合しαリングが完成する.Pba3とPba4が存在しないとα4サブユニットの取り込みに失敗し,α2が2コピー取り込まれた異常な組成のαリングが形成しCP形成が止まってしまう106).Pba1–Pba2ヘテロ二量体はHbYXモチーフを持ちαリングのRP結合面に蓋をするように結合することでαリングを安定化させる(図4)107,108).次にαリングをテンプレートとしてβ2,β3,β4が取り込まれる.Pba3–Pba4はβサブユニットと似た構造を持ちβリング側に結合しているが,β4が取り込まれる際に立体障害により前駆体から排除される109).次に,Ump1とβ5,β6,β1が結合し15S前駆体を形成する.なお,βサブユニットはプロペプチドが付加した前駆体として翻訳され(正確にはプロテアーゼ活性を持つβ1,β2,β5を含む5種類),前駆体のまま取り込まれる110).Ump1は天然変性タンパク質でありβリング中央に存在する.15S前駆体は比較的安定な中間体であるが,β7が結合しハーフCPとなると速やかに二量体化しCP構造をとる109).Ump1やβサブユニットのプロペプチドはこの二量体化に重要であり,CP構造をとるとプロペプチドが切断されCPは活性化し,Ump1は新生CPの最初の基質として分解される.CPが完成するとαリングの構造変化によりPba1–Pba2は解離する111).
ヒトのCP形成機構も酵母とほぼ同様であるが,POMP(ヒトUmp1)がαリングに結合するタイミングや,PAC1–PAC2(ヒトPba1–Pba2)が新生CPにより分解されるなど,細かい点は異なる101,107,112).CP形成はPOMPの量が律速となっており,POMPを過剰発現するとCP量が増加する113).ごく最近,POMPの発現がマイクロRNAのmiR-101により調節されていることが明らかとなった99).miR-101はPOMPの発現を抑制するが,ある種のがん細胞ではmiR-101が減少しており,その結果,CP量およびプロテアソーム量が増加する.
ⅱ)RPの形成機構
RPの構成サブユニット変異体を用いた解析より,基底部と蓋部は別々に形成することがわかっている114).筆者らは機能未知のPIPsであるNas6について解析したところ,Nas6が完成したプロテアソームには存在せずフリーのRPや基底部(およびその前駆体)にのみ存在することを見出した.さらに同様の特徴を持つPIPsとしてNas2,Rpn14,Hsm3を同定し,これらが基底部の分子集合を支援するシャペロンであることを解明した(これらRPシャペロンは同じ時期に複数の研究室から報告され,驚いたことを記憶している)19,115–117).
先に述べたとおり基底部のATPaseサブユニットは二つずつのペアを組むが,四つのRPシャペロンは異なるRptのC末端領域と選択的に結合し,Nas6–Rpt3–Rpt6–Rpn14(Nas6モジュール),Hsm3–Rpt1–Rpt2–Rpn1(Hsm3モジュール),Nas2–Rpt4–Rpt5(Nas2モジュール)の三つの中間体を形成する.これら四つのRPシャペロンはすべて遺伝子破壊しても致死とはならないが,プロテアソーム量が約10%まで減少し,熱ストレスやタンパク質毒性ストレスに超感受性を示した19).RPシャペロンはそれぞれ異なるドメイン構造を持つが(Nas6はAnkyrinリピート,Rpn14はWD40リピート,Hsm3はHEATリピート,Nas2はPDZドメインを持つ),筆者らはRpn14,Hsm3,Nas2の結晶構造解析および機能解析を行い,RPシャペロンがパートナーのATPaseドメインを正確に識別する機構と基底部形成の順番を明らかにした118–120).遺伝学的にNas6モジュール形成が最も重要なステップであるが,まずNas6モジュールにNas2モジュールとRpn2–Rpn13が結合し,最後にHsm3モジュールが結合することで基底部が完成する.このとき,Nas6,Rpn14,Hsm3は基底部に結合したままだが,Nas2はHsm3モジュールの結合の際にRpt1により押し出され解離することがわかった.一方,脱ユビキチン化酵素Ubp6がRP前駆体と相互作用し,形成途中のRPにユビキチン化基質が結合するのを防いでいることを明らかにした9).
最近,Adc17が酵母の遺伝学スクリーニングにより新しいRPシャペロンとして同定された103).Adc17はRpt6のN末端領域に結合し,Rpt6のフォールディングを補助することでRpt3–Rpt6二量体形成を促す.他の四つのRPシャペロンと異なり高等真核生物には保存されていないが,Adc17は小胞体ストレスやアミノ酸アナログによって強く誘導され,ストレス応答時のプロテアソーム量の増加に必須である.これはRpt6を含むNas6モジュール形成がRP形成の律速段階であるためと考えられている.また,不思議なことにAdc17を含めてほとんどのプロテアソームシャペロンはPACE配列を持たない91,103).そのため,Rpn4以外の転写因子によって発現が制御されている可能性が高い.
基底部の形成はCPに非依存であると考えられているが,CPのαリングをテンプレートとしたCP依存的経路も報告されている121,122).このCPテンプレートモデルは基底部中間体内のRpt6のC末端がCPのα2–α3間のポケットと結合し,それを起点として各Rptサブユニットの配置が決定されるというものである.このとき,RPシャペロンはRpt6が正しい位置に結合するのを支援するらしい.しかし,基底部中間体–CP複合体の存在が確認されていないのが欠点である.
一方,蓋部形成に関与する専用シャペロンについては今のところ決定的なものは見つかっていない.筆者らは蓋部サブユニット変異体で生じる部分集合体の解析を行った.その結果,まずRpn3–Rpn7–Rpn15とRpn5–Rpn6–Rpn8–Rpn9–Rpn11という二つのモジュールが形成し,それらが会合した後にRpn12が取り込まれ蓋部が完成することが示唆された(図4)123).その後,試験管内の再構築実験によりこの経路が実証され,蓋部が完成して初めてプロテアソームに取り込まれることが明らかとなった124).なお,既知のプロテアソーム専用シャペロンはαリングやATPaseリングのように相同性の高いサブユニットを正確な位置に配置させるために存在するとされる.一方,蓋部の構成サブユニットは相同性が低いため自発的に複合体を形成できると考えられている.
その後,蓋部と基底部は会合し,Rpn10が取り込まれることでRPが完成する124).
ヒトのRPも酵母とほぼ同様に形成すると考えられているが,基底部形成においてAdc17ホモログが存在しない点や,Nas2モジュールが最後に取り込まれる点で異なる116).また,酵母のRP形成はAdc17が重要であるが,ヒトでは後述のようにRpn6の発現が律速となる98,103).
ⅲ)分子シャペロン
プロテアソーム専用シャペロン以外にも,いわゆる一般的な分子シャペロンもプロテアソーム形成に関与している.Hsp90は熱ストレス時のプロテアソームレベルの維持に必要であり,蓋部の形成に関与している125).また,テイルアンカー型膜タンパク質の膜挿入を制御するTRC(transmembrane recognition complex)経路がCP形成に関与することが報告されている126).TRC経路は形成途中のCPを小胞体膜上に局在化させることによりCP形成を促進させるらしい.さらに異常膜タンパク質の分解に関与するBAG6がRP形成に必要であり,特にRpt4–Rpt5–p27複合体形成を促進させることが報告されている126).これらの分子シャペロンが直接プロテアソーム形成に関与しているのかは不明であるが,ハウスキーピング分子同士の制御機構であり興味深い.
3)プロテアソーム安定化因子と解離因子
プロテアソームの安定性を制御する因子としてEcm29(ヒトではKIAA0368)が知られている.Ecm29はHEATリピートを持つ約200 kDaのタンパク質で主要なPIPsとして同定された.ecm29破壊株ではプロテアソームが不安定化しRPとCPに解離しやすくなること,Ecm29はRPとCPのどちらとも相互作用できることから,Ecm29はRPとCPをつなぎとめるベルトのような役割を果たしていると考えられる7,73).一方で,Ecm29はプロテアソームの解離因子としても知られており,酸化ストレス時にプロテアソームが損傷を受けるとRPとCPに解離させる機能を併せもつ127).これは相反する機能に思えるが,Ecm29は異常な構造のプロテアソームと相互作用するという報告もあり,プロテアソームの品質管理を実行する分子だとすると説明がつく128–130).なお,哺乳動物細胞ではEcm29はプロテアソームをエンドソームに局在化させるアダプタータンパク質でもある131).
さらにヒトではプロテアソームの解離因子としてUBLドメインを持つホスファターゼUBLCP1が知られている132).機能はあまりわかっていないがプロテアソームの多くのサブユニットは細胞内で高度にリン酸化されている.UBLCP1は試験管内でホスファターゼ活性依存的にプロテアソームをRPとCPに解離させること,UBLCP1をノックダウンすると核内のプロテアソーム量が増加することから,UBLCP1は核内プロテアソームの解離因子として何らかの機能を持つとされる.
4)プロテアソームの分解
プロテアソームは長寿命のタンパク質複合体であるが,酸化ストレスなどにより損傷を受けたプロテアソームは細胞にとって有害であることが強く想定される.ごく最近,機能が低下したプロテアソームがオートファジーにより選択的に分解されることがシロイヌナズナで報告された(proteaphagyと命名)132).Proteaphagyは窒素源枯渇のほか,プロテアソーム阻害剤処理時やプロテアソーム変異体で観察される.このとき,プロテアソームは複数のサブユニットが高度にユビキチン化されており,ユビキチン選択的オートファジーを受けていることが示された.意外なことにproteaphagyのオートファジー受容体はプロテアソームサブユニットのRpn10であった.シロイヌナズナのRpn10は三つのUIMを持つが2番目のUIMでATG8(ヒトではLC3)と直接相互作用できる.ヒトや酵母はこの二つ目のUIMを持たず,LC3やATG8とは相互作用しないようであるが,ヒトや酵母でproteaphagyが起こるのかどうかを含めて今後の解析が待たれる.
5)生理的なプロテアソームレベルの変動
ここまでプロテアソームレベルの調節機構を紹介してきたが,最近の研究により,生理的な条件下においてプロテアソームレベルが大きく変動すること,個体の寿命にも関与することがわかってきた.
ⅰ)細胞分化に伴うプロテアソーム量の減少
プロテアソームレベルは細胞の種類によって異なるが,特にES細胞ではプロテアソームの分解キャパシティーが亢進していることが知られている98,133).ES細胞はタンパク質毒性ストレスに脆弱であり高いプロテアソーム活性が全能性の維持に必要であるらしい.興味深いことにES細胞の分化を誘導するとプロテアソーム活性が徐々に減少する.これは量的な制御であり,たった一つのRPサブユニットRpn6(PSMD11)の発現レベルに依存していた98).出芽酵母ではすべてのプロテアソームサブユニットはほぼ完成したプロテアソームに取り込まれている(後述)が,ヒトの多くの細胞では,RP前駆体やフリーのCPが余剰に存在しており,Rpn6の取り込みがプロテアソーム形成の律速となっているようである.実際,分化後の細胞にRpn6を過剰発現させるとプロテアソーム量が増加した.Rpn6の転写はフォークヘッド転写因子のFOXO4によって制御されており,分化後のES細胞ではFOXO4が減少すること,Rpn6のみがFOXO4により制御されることが示されている.分化後はプロテアソーム量が減少するが,なぜプロテアソーム量を減じる必要があるのだろうか? プロテアソームは基質非存在下ではATPase活性が低いので単にエネルギーのむだ使いを防いでいるためとは考えにくい.
ⅱ)プロテアソームレベルと寿命
ES細胞にみられたFOXO型転写因子とRpn6によるプロテアソームレベルの調節は線虫においても保存されている134).線虫のFOXO型転写因子DAF-16は飢餓や高温などのストレスにより寿命延長やストレス応答に関わる遺伝子を発現させるが,さらにRpn6の発現を介してプロテアソームレベルを制御していることが明らかとなった.興味深いことにRpn6の強制発現により線虫の寿命が約2倍に延長した.一方,ショウジョウバエでは加齢に伴いプロテアソーム活性が減少すること,Rpn6ではなくRpn11の過剰発現により個体の寿命が延長することが示されている135).これらの結果より,プロテアソームレベルが個体の寿命に大きな影響を与えることが明確となった.
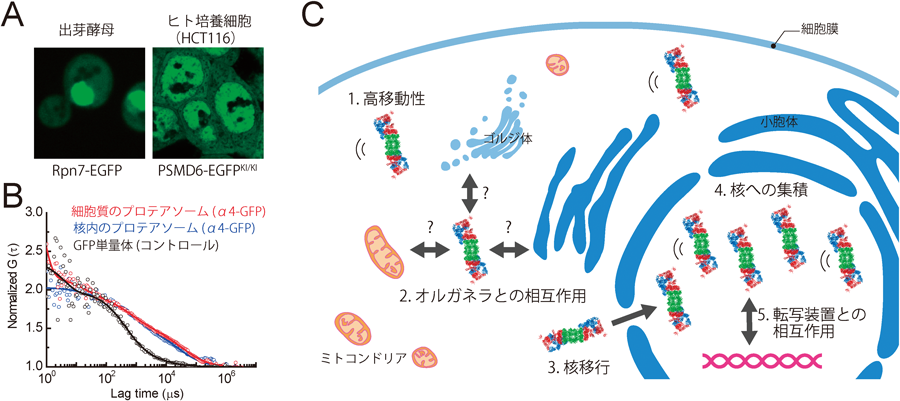
異常タンパク質の大部分は翻訳時に産生するため,細胞は品質管理機構としてRQC(ribosome quality control)およびERAD経路を持つ.いずれの経路でも細胞質のプロテアソームが異常タンパク質のクリアランスの最終段階を担っている136,137).そのため,プロテアソームは細胞質に専ら存在すると思われがちであるが,酵母やがん細胞など増殖の盛んな細胞ではプロテアソームは主に核に存在する(図5).核内のプロテアソームはユビキチン系とともに転写調節やDNA修復,クロマチン構造変換,mRNAの核外輸送に関与するとされるが,詳細な分子機構はあまりよくわかっていない138–141).少なくとも酵母ではプロテアソームを核から排除すると大量にユビキチン化タンパク質が蓄積することから,核内タンパク質の品質管理や転写因子の分解を行っていると考えられる142).一方,プロテアソームは基本的には細胞質や核質に一様に観察されるが,タンパク質毒性ストレスなどによりアグリソーム(aggresome)や核内fociに局在化することが報告されている.ここではまず,筆者らの蛍光相関分光法によるプロテアソーム動態解析を紹介した後,クライオ電子線トモグラフィーによる細胞内プロテアソームの直接観察,そしてプロテアソームを含む様々な細胞内構造体を紹介する.
1)蛍光相関分光法を用いた動態解析
プロテアソームは基本的には細胞質や核質に一様に観察され,かつ拡散性であるため,各プロテアソームサブユニットに対する抗体を用いた多重染色や2色の蛍光タンパク質タグを用いても複合体としてのプロテアソームの機能を言及するのは困難である.そこで我々は,蛍光相関分光法(fluorescence correlation spectroscopy: FCS)を用いてプロテアソームの細胞内動態を解析している143).FCSは蛍光標識した対象分子の微小空間における蛍光のゆらぎを計測することで,対象分子の濃度と拡散係数を決定する手法であり,拡散係数より分子の形状や分子量を決定できる(図5)139,144).さらに2色の異なる蛍光タグを用いたFCSは蛍光相互相関分光法(fluorescence cross-correlation spectroscopy: FCCS)と呼ばれ,二つの異なるタンパク質間の相互作用を生きた細胞で計測することが可能である.まず,出芽酵母プロテアソームのさまざまなサブユニットに緑色蛍光タンパク質(GFP)を融合した株を作製し,プロテアソーム機能が完全に保持されているものについて生細胞FCS解析を行った.その結果,プロテアソームのサブ複合体CP,蓋部,基底部の各構成サブユニットはいずれもよく似た動態を持つこと,細胞質および核においてそれぞれ2種類の成分を持つことがわかった.一つは見かけ分子量1~10 Mの速い成分であり,プロテアソーム形成に欠損を示すRP変異体において拡散がさらに速くなったことから完成型のプロテアソームそのものであることがわかった.もう一つは見かけ分子量32 Gの遅い拡散を持つ成分であり,この値はプロテアソーム10,000分子以上に相当する.顕微鏡下でそのような集合体は観察されないため,プロテアソームが何らかの細胞小器官(オルガネラ)と相互作用していることが想定された.また,この遅い拡散成分はプロテアソーム阻害剤処理により減少したため,ユビキチン化タンパク質を介したものであることが示唆された(未発表).核内については,RNAポリメラーゼⅡを阻害すると遅い成分が減少したことから,核内のプロテアソームは転写装置と相互作用していることがわかった.次に細胞内濃度を決定したところ,CP,蓋部,基底部はいずれも細胞質では約200 nM,核では約1 µMであり,計算すると酵母1細胞あたりプロテアソームは約10,000分子存在(核と細胞質でそれぞれ約5000分子ずつ存在)することがわかった.これはリボソームの1/10~1/20の分子数に相当する.なお,筆者らの決定した値はMannらの質量分析計による測定結果とよく一致した145).
プロテアソームの複合体形成率を測定するため,RPサブユニットにGFPを,CPサブユニットにmCherryを融合した株を作製しFCCS測定を行った.その結果,驚いたことに細胞質においても核においてもRPとCPの複合体形成率は90%以上であり,かつ構成サブユニットはほぼすべてプロテアソーム中に存在することが明らかとなった.この結果は意外であったが,従来の生化学的解析で大量に存在するとされたフリーサブユニットやサブ複合体がnative電気泳動などの迅速な分画法ではほとんど検出されないという結果と一致する105).
酵母は分裂期に核膜が崩壊しないため,プロテアソームが核に局在化するためには分子集合のどこかの段階で核膜孔を通過する必要がある.プロテアソームは巨大分子であるため形成中間体の段階で核に移行し,核内で完成するというモデルが主流となっていた146,147).出芽酵母インポーティンαのさまざまな変異体のうちsrp1-49温度感受性変異株はプロテアソームの核移行に欠損を示すアリルであり,制限温度下ではプロテアソームサブユニットは細胞質および核膜の外側に蓄積する148).そこでsrp1-49変異体のプロテアソームのFCCS解析を行ったところ,野生株と変異株とでは複合体形成率に差はみられなかった.つまり,プロテアソーム形成と核移行は共役していないことが示唆された.
最後に,プロテアソームが物理的に核膜孔を通過できるか,RPサブユニット(Rpt1)とCPサブユニット(α4)をつないだ安定化型プロテアソーム変異体を作製し解析した.この安定化型プロテアソームはRPとCPに解離せず,野生型のプロテアソームと同等の機能を保持しているが,野生型と同様に核に局在化できることがわかった.つまり,プロテアソームは完成体として核膜孔を通過できることが示唆された.核膜孔の内径は最大39 nmまで拡大することから,プロテアソーム(25 nm×45 nm)は長軸方向でなら核膜孔を通過できると考えられる143,149,150).しかし,核膜孔の内側は非常に疎水性が高いため,輸送途中のプロテアソームには何らかの保護分子が結合している可能性がある.後述の通り,酵母プロテアソームはグルコース濃度に依存し,細胞質と核間をダイナミックに行き来するので,巨大分子の細胞質核間輸送のモデル分子となることが期待される.
2)クライオ電子線トモグラフィーによるプロテアソームの直接観察
細胞内のプロテアソームの形状を直接可視化できないだろうか? Baumeisterらのグループはこれまで粘菌のリボソームやプロテアソーム,アクチン繊維などをクライオ電子線トモグラフィーにより観察してきたが151),ごく最近,ラット海馬由来神経細胞の中のプロテアソームを35~50 Åの分解能で可視化することに成功した152).クライオ電子線トモグラフィーによる観察は厚みのある試料では難しいため,細胞質のみの観察ではあるが,まずプロテアソームは約200 nMの濃度で存在すること,30Sと26Sのプロテアソームは約3 : 7の比で存在することを明らかにした(つまりシングルキャップのプロテアソームが多い).さらに,待機状態と活性化状態のプロテアソームを識別することが可能であり,驚いたことに約80%のプロテアソームが待機状態であった.つまり,ストレスのない状態ではわずか20%のプロテアソームしか使われておらず,プロテアソームの分解キャパシティーは十分な余力が残されていることが明らかとなった152).残念ながらストレス存在下や核内のプロテアソームについては今後の報告を待たなければならないが,インタクトな細胞でプロテアソーム動態を可視化した画期的な論文である.
3)プロテアソームを含む細胞内構造体
パーキンソン病やアルツハイマー病,筋萎縮性側索硬化症,ハンチントン病などの神経変性疾患ではユビキチン陽性の封入体が観察される153).これらの疾病の原因となる凝集性タンパク質を発現させると,プロテアソームは細胞内のさまざまなコンパートメントでfociを形成することがわかってきた.これらのfociを解析することで封入体形成の初期過程を理解できると考えられる.ここでは細胞質と核に形成されるプロテアソームfociを紹介するとともに休止期の酵母でみられるプロテアソーム顆粒を紹介する.
ⅰ)JUNQとアグリソーム
出芽酵母に易凝集性タンパク質を発現させると,JUNQ(juxta nuclear quality control compartment)またはIPOD(insoluble protein deposit)と呼ばれる細胞内コンパートメントが形成される154).JUNQにはユビキチンとプロテアソームが集積しており,プロテアソーム変異体やプロテアソーム阻害剤でJUNQの形成が顕著に誘導されることから,JUNQは比較的溶解度の高い凝集タンパク質がユビキチン化された後に運ばれプロテアソームによって分解されるコンパートメントとされる.一方,IPODはpolyQタンパク質などのアミロイドを形成する不溶性タンパク質が集積する液胞近傍のコンパートメントである.IPODにはオートファジーマーカーAtg8(酵母のLC3)が存在するためオートファジーによるクリアランスに関与するとされる.ごく最近,ヒトの細胞でも易凝集性タンパク質の過剰発現によりJUNQが形成されることが報告された155).ヒトの細胞では以前より核近傍に観察される封入体としてアグリソームが知られているが,JUNQに運ばれたタンパク質がプロテアソームで分解されるのに対しアグリソームに運ばれるタンパク質はオートファジーで分解される点で異なる155,156).しかし,アグリソームとJUNQは共通点が多く,ともにプロテアソーム阻害剤により誘導され,プロテアソームやユビキチンが集積すること,双方とも中間径フィラメントのビメンチンに包まれた構造物であることから,JUNQはアグリソームの前駆体ではないかと考えられている155,157).なお,JUNQもアグリソームも細胞内の核近傍に一つだけ存在するが,これは脱アセチル化酵素HDAC6が凝集タンパク質とダイニンのアダプターとして働き,微小管上を逆行輸送された結果である.興味深いことに,JUNQは細胞分裂に伴い一つの娘細胞に不均等分配される.そのため,ヒトの細胞においても出芽酵母と同様に細胞分裂に伴う細胞の若返り機構があるのではないかと提唱されている.
ⅱ)ClastosomeとPMLボディー
核内におけるタンパク質の品質管理機構はまだよくわかっていないが,酵母ではSan1とUbr1が核内変性タンパク質のユビキチンリガーゼとして同定されている158,159).また,細胞質の易凝集性タンパク質はHsp70のコシャペロンSis1により核に輸送されプロテアソームで分解されるという報告がなされ,核内におけるプロテアソーム分解が注目されている160).先に述べたように核内のプロテアソームは核小体を除き一様に存在しているが,動物細胞ではユビキチンとプロテアソームを含む1~3個のfociがまれに観察され,Clastsome(ギリシア語の分解を意味するKlastosに由来)と名づけられている.Clastosomeはプロテアソーム基質と共局在することから,核内のタンパク質分解の場ではないかと考えられている161).
PMLボディーは直径0.2~1 µmの球状の構造体で転写やDNA修復,タンパク質分解などさまざまな機能を持つ核内構造物である162).PML自身もSUMO化依存的にユビキチン化されプロテアソーム分解を受けるが,特に,PMLの六つのアイソフォームのうちPML IVはpolyQタンパク質(変異ATNX7)の分解に関与すること,ユビキチンやプロテアソームが高度に集積することが以前より知られていた163).最近,PML IVがpolyQタンパク質のみならず様々なミスフォールドタンパク質を認識するSUMOリガーゼであること(SUMO化の後,ユビキチン化とプロテアソーム分解が起こる),核内タンパク質の品質管理を担っていることが報告された164).
ⅲ)プロテアソーム貯蔵顆粒
酵母はグルコースを使いつくし定常期に入ると,プロテアソームが核から細胞質に移動し,細胞質に1~2個の顆粒構造を形成する165).このプロテアソーム貯蔵顆粒(proteasome storage granules: PSGs)は可逆的な構造体であり,グルコースを培地に添加すると数分で消失する143).PSGsの構成成分や生理的な意義はまったく不明であるが,プロテアソームを顆粒内に隔離することでプロテアソームを酸化ストレスなどから保護し,再び増殖期に戻る際にプロテアソームレベルを速やかに回復させる機能をもつのではないかと考えられている.形成機構としては細胞内pHの低下がトリガーになることが示唆されている166).PSGsが動物細胞においても形成されるかは不明である.