細胞内ではシグナル伝達によって,分子間の相互作用,翻訳後修飾,タンパク質の構造変化や酵素活性の変化が秩序正しく営まれている.この秩序が時空間的に厳密に制御されること,すなわち細胞内の「いつ,どこで」分子の活性化が生じるかが,遺伝子発現制御や細胞の形態変化などの表現型を決めている.たとえば,神経成長因子受容体は局在の違いにより異なる下流因子を活性化することがわかっており,細胞膜での活性化が増殖に,エンドソームでの活性化が細胞の分化に重要であると考えられている1,2).ほかにも,エンドソームから発信されるシグナルの重要性が徐々に解き明かされつつある3,4).すなわち,エンドソームは単なる物質を運ぶオルガネラではなく,自ら積極的にシグナルを発するプラットフォームとしても注目されつつある.
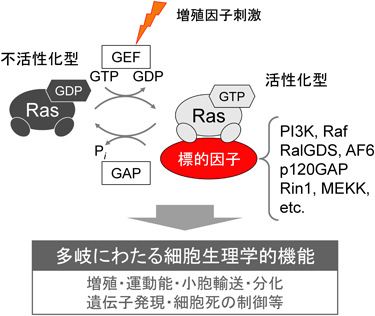
低分子量GTP結合タンパク質であるRasは,シグナル伝達において多機能かつ中心的な役割を担っているにも関わらず,酵素活性としてはGTPの加水分解能を有するのみである.したがって,下流の標的因子群を「いつ,どこで」活性化するかを精密に制御する機構が存在し,多彩な役割を如才なく担うことができるのではないかと推察されている(図1).
本稿では,蛍光バイオイメージングを用いたRasシグナルの時空間的解析により明らかとなった,エンドソームから発信されるRas-PI3K(phosphoinositide 3-kinase)シグナルの意義と,外来因子取り込みに関与する細胞内シグナルネットワークについて概説する.
1)Rasシグナル
Rasは細胞の増殖,分化,運動能,遺伝子発現のほか,細胞死の制御や小胞輸送など数多くの基本的な細胞機能を制御する低分子量GTP結合タンパク質である5–7)(図1).Rasはグアニンヌクレオチド交換因子(guanine nucleotide exchange factor: GEF)によってグアノシン三リン酸(guanosine triphosphate: GTP)が結合した活性型に変換され,Ras結合ドメイン(Ras binding domain: RBD)を介して標的因子と結合し下流にシグナルを伝える.Rasを活性化する経路として,アダプター分子Grb2とRasGEFのSos(son of sevenless)を介した増殖因子刺激–チロシンリン酸化の経路と8),カルシウムシグナルなどセカンドメッセンジャーを介する経路9)が知られている.標的因子と乖離した後のRasは,GTPase活性化タンパク質(GTPase-activating protein: GAP)により自身のGTPase活性が亢進し,結合しているGTPを加水分解してグアノシン二リン酸(guanosine diphosphate: GDP)が結合した不活性型となる.したがって,Rasは活性型と不活性型に可逆的に変化することで分子スイッチとして機能しているといえる.
代表的なRasの標的因子としては,c-Raf1,Ral guanine nucleotide dissociation stimulator(RalGDS),ホスホイノシチド3-キナーゼ(phosphoinositide 3-kinase: PI3K)などが知られている.c-Raf1はextracellular signal-regulated kinase/mitogen activated protein kinase(ERK/MAPK)経路を介して遺伝子発現のシグナルを,RalGDSはRalの活性化を介して小胞輸送のシグナルを,PI3KはAktのリン酸化を介して生存のシグナルをそれぞれ伝達している.
RasファミリーはC末端側にCAAXモチーフと呼ばれる膜局在化ドメインを有しており,古くは細胞膜のみで働くと考えられてきた.しかし,2000年ころからRasファミリーが時空間的に局在と活性を制御されていることを示す例が多数報告されてきた.松田博士らのグループは,増殖因子刺激によりRasが細胞辺縁部の細胞膜で活性化し,一方同じく低分子量GTP結合タンパク質Rap1が主に細胞の中心部で活性化することを報告した10).このような活性化がGAPによって空間的に制御されていることも報告された11).また,エンドサイトーシスを阻害した際には,Rap1の活性化のみが抑制される10).さらに,神経成長因子(nerve growth factor: NGF)によるRap1の活性化はNGF受容体であるTrkAのエンドサイトーシスに依存するが,Rasの活性化は依存しないという報告12)と併せて,増殖因子刺激シグナルをRasとRap1がエンドサイトーシスを用いて使い分けることが明らかとなってきた.ほかにもMark Phillips博士らのグループから,Rasは細胞膜だけでなくゴルジ装置や小胞体にも局在し,ERKの活性化を介して線維芽細胞の悪性形質転換(がん化)を促進することが報告された1,2,13,14).この場合,細胞膜と細胞小器官のRasは時間的に異なる活性化動態を示し,細胞内カルシウムが重要な役割を担う.たとえば細胞膜での活性化は一過性でゴルジ装置での活性化は遷延するが,これはカルシウム依存性RasGAPであるCAPRIが細胞膜で特異的に機能するためである15).さらに,Rasがエンドソームにも局在することも報告されており3–5,16,17),エンドソームに局在するRasの機能や役割にも注目が集まっている.しかし,Ras自身の時空間制御に関する研究の進展に対し,Rasの下流因子活性化の時空間制御に関しては未知の部分が多い.
2)PI3K
PI3Kは真核生物の細胞膜構成成分の一つであるイノシトールリン脂質のイノシトール環3位の水酸基をリン酸化する酵素であり,基質に応じてホスファチジルイノシトール3-リン酸[phosphatidylinositol 3-phosphate: PtdIns(3)P],ホスファチジルイノシトール3,4-ビスリン酸[PtdIns(3,4)P2],PtdIns(3,5)2,ホスファチジルイノシトール3,4,5-トリスリン酸[PtdIns(3,4,5)P3]が生成される.構造によりクラスⅠ,Ⅱ,Ⅲに分類され,クラスⅠがもっぱらPtdIns(4,5)P2をリン酸化することでPtdIns(3,4,5)P3を産生するのに対し,クラスⅡはPtdInsとPtdIns(4)Pに対する基質特異性が高く,クラスⅢのPI3KはPtdInsからPtdIns(3)Pを産生する18).RBDはクラスⅠとクラスⅡに存在するが,その活性制御と機能が最も研究されているのはクラスⅠである.
PI3Kは,その下流因子の活性化を介して細胞分化・増殖や代謝,細胞遊走,細胞骨格の再構築など多様な生物活性を引き起こす.中でもAktはPHドメインを介してクラスⅠのPI3Kが産生したPtdIns(3,4,5)P3により細胞膜にリクルートされて活性化し,細胞の生存やインスリンの分泌などさまざまな生理機能に関与する5–7,19).ほかにPXドメインやFYVEドメイン[主にPI(3)Pに結合]などの配列を持つタンパク質が,クラスⅡやクラスⅢのPI3Kの下流因子として機能する.
これまでのPI3Kの研究は阻害剤もしくはノックアウトマウスを用いた解析が主であったため,その制御機構とりわけRasによるPI3Kの活性制御については議論の余地が多い.特にクラスⅠのPI3Kの制御サブユニットはSrc homology 2(SH2)ドメインを有しているため,チロシンキナーゼの下流におけるいくつかの機能においてはRas非依存性であると報告されている8,20).しかし近年,PI3KのRBDにRasと結合できない点突然変異を導入したノックインマウスを用いた研究から,RasとPI3Kの結合がRas依存的な腫瘍形成に必須であるとの報告があり21),Rasの標的因子としてのPI3Kが再注目されるとともに,上流因子と機能の特異性の決定メカニズムの解明が待たれるところである.
3)エンドソームから発信されるRas-PI3Kシグナル
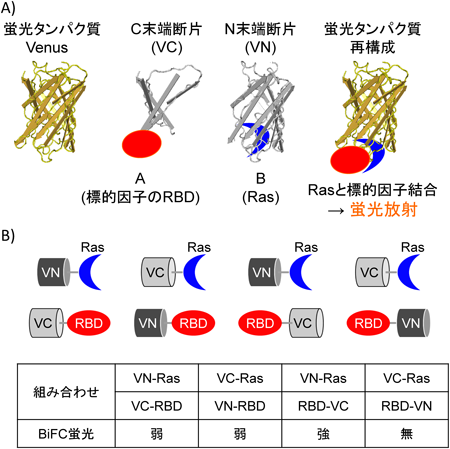
筆者らはRasと標的分子の複合体の挙動を時空間的に解析するために,2分子間の相互作用を蛍光強度の増加としてモニターできる蛍光タンパク質再構成法(bimolecular fluorescent complementation: BiFC)22,23)を用いた.BiFC法の原理を図2に示す.蛍光タンパク質をN末端とC末端側に分断(図では172-173アミノ酸)した場合,どちらも蛍光は発しない.しかし,それぞれの断片に結合したタンパク質の相互作用に応じて蛍光タンパク質が再構成されると蛍光を発する.すなわち,酵母ツーハイブリッド等で使われるGal4再構成系の蛍光タンパク質版で,タンパク質間相互作用が蛍光強度の増加として検出できる.
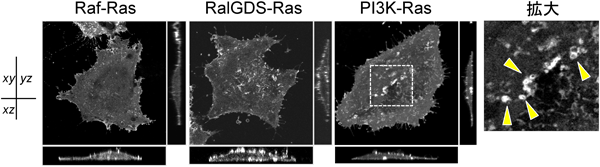
恒常的活性型RasのN末端側に蛍光タンパク質YFPのN末端断片を,標的因子として有名なRaf,RalGDS,PI3KのC末端側にYFPのC末端断片をそれぞれ融合した組換えタンパク質をCOS-1細胞に発現させたところ,すべての組み合わせにおいて細胞膜からの蛍光が観察された(図3).すなわち,これまでの報告どおりこれらの因子は細胞膜でRasと結合し,活性化されることが確認された.しかし,RafとRasおよびRalGDSとRasの結合は細胞膜からのみ蛍光が観察されたのに対し,PI3K-Ras複合体は細胞膜だけでなく細胞質の小胞状構造にも局在することがわかった(図3).さらに,PI3KのRBDと野生型Rasを発現したCOS-1細胞を上皮増殖因子(epidermal growth factor: EGF)で刺激し,蛍光顕微鏡によりその挙動をリアルタイムでより詳細に解析した.EGF刺激後10分経過したあたりから,Ras-PI3K複合体由来の蛍光強度の上昇が観察され,さらにRas-PI3K複合体が細胞膜から小胞状構造に移行するようすを捉えることに成功した.この構造は初期エンドソームマーカーであるearly endosomal antigen(EEA1)と共局在することから,Ras-PI3K複合体は初期エンドソームに局在することが示された.実際,エンドサイトーシスの抑制下において,EGF刺激による細胞膜でのRas-PI3Kの複合体形成は認められたが,エンドソームへの局在はみられなくなった.
さらに,EGF刺激依存的にPI3Kの反応生成物であるPtdIns(3,4,5)P3が細胞膜だけでなくエンドソームにおいても検出されたことから,Ras-PI3Kシグナルがエンドソーム上でも活性化していることが示唆された.興味深いことに,ドミナントネガティブ型Rasを発現すると,EGF刺激依存的なPtdIns(3,4,5)P3の産生は細胞膜のみで検出され,エンドソーム上では検出されなかった.以上から,細胞膜でのPI3Kの活性化にRasは必須ではないが,エンドソーム上での活性化にはRasが必要であることが示された24).言い換えれば,これまで議論のあったRasによるPI3Kの活性制御について,エンドソーム上での活性化には,Rasが上流因子として機能することが明らかとなった.また,クラスⅢのPI3KであるVPS34(vacuolar protein sorting 34)がエンドソーム上でPtdIns(3)Pを産生することは報告されていたが25,26),クラスⅠのPI3Kがエンドソーム上でPtdIns(3,4,5)P3を産生しているか否かは未知であった19).筆者らの論文発表の後24),PtdIns(3,4,5)P3がエンドソーム上で産生され,ファゴソームの形成に関与することを示す報告もあり27),筆者らはエンドソームから発信されるRas-PI3Kシグナルの重要性を再認識するとともに,エンドサイトーシス制御との関連に注目した.
1)エンドサイトーシス
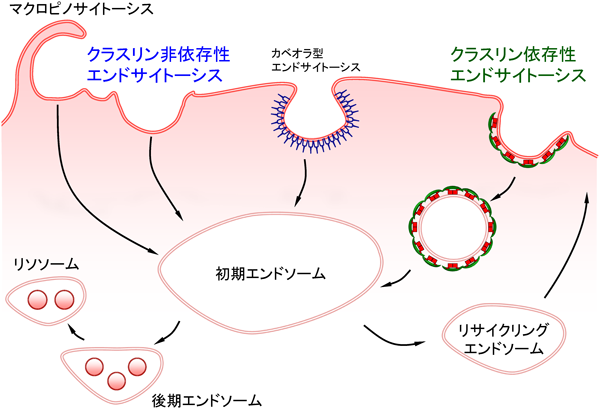
エンドサイトーシスは細胞外物質を細胞内に取り込むための機構であり,細胞が環境変化に応答するために必要である.エンドソーム膜の構成成分やその形成機構で分類されており,古くからよく研究されているクラスリン依存性エンドサイトーシス,カベオラ型エンドサイトーシス,最近注目を集めつつあるクラスリン非依存性エンドサイトーシス等があげられる10,28)(図4).
クラスリン依存性エンドサイトーシスは受容体依存性エンドサイトーシスとも呼ばれ,主にリガンドと結合した受容体が細胞内に取り込まれるための機構である.リガンドと受容体が相互作用すると,クラスリンが受容体周囲の細胞内膜に集積してクラスリン被覆を形成する.それに伴い細胞膜が陥入し,細胞膜のくびれの部分をGTP結合タンパク質ダイナミンが切断することで,クラスリン被覆小胞として細胞膜から脱離し細胞内に取り込まれる.小胞は脱被覆化した後に融合して初期エンドソームとなり,一部は後期エンドソーム,リソソームへと進む分解経路,一部はリサイクリングエンドソームへと進む再利用経路へと運命を分かつ29).
カベオラ型エンドサイトーシスは,細胞膜表面のカベオラと呼ばれるくぼみ構造を介して物質の取り込みを行う.カベオラはスフィンゴ脂質やコレステロールに富んでおり,コレステロールに結合して自己会合するカベオリンによってくぼみが形成される.その後,クラスリン依存性エンドサイトーシスと同様にダイナミンによって細胞膜先端部が癒合し,最終的に小胞を形成し細胞内に取り込まれる30).
クラスリン非依存性エンドサイトーシスは,クラスリンやカベオラ依存性エンドサイトーシスとは異なり被覆小胞を形成せずに物質を取り込む様式をとり,近年になって盛んに研究が行われている.低分子量GTP結合タンパク質であるRhoファミリーが制御するエンドサイトーシス,死細胞や細菌などを取り込むファゴサイトーシスや,マクロピノサイトーシスと呼ばれる細胞外液ごと一気に飲み込んでしまう機構が知られている31).
PI3Kに関してはクラスⅢのPI3KであるVPS34がオートファジー,エンドサイトーシス,ファゴサイトーシスにおける小胞輸送に関わることが知られている.たとえばファゴサイトーシスにおいては,VPS34が産生したPtdIns(3)Pがエンドソームに集積することで,ファゴソームの形成が促進されると考えられている19).また,マクロピノサイトーシスにおいては細胞膜で産生されたPtdInd(3,4,5)P3によりマクロピノソームの形成が促進されると考えられている32).ほかにも最近,クラスⅡのPI3KであるPI3KC2αがPtdIns(3,4)P2を産生することで,クラスリン被覆ピットの形成を介してクラスリン依存性エンドサイトーシスを制御することが報告された33).一方,Rasに関しては,Rab5のGEFであるRIN1に直接結合し,Rab5の活性制御を介してエンドサイトーシスを促進することが知られている9,34,35).また,近年ではH-Rasがマクロピノソームの成熟化に寄与することも報告されている36).
2)Ras-PI3Kシグナルによるエンドサイトーシスの制御
PI3KがRas依存的にエンドソームに局在し,エンドソームからシグナルを発信することがエンドサイトーシス制御に関与するか検討するために,PI3Kγをノックアウトしたマウス胎仔線維芽細胞(murine embryonic fibroblast: MEF)37)を用いてクラスリン依存性エンドサイトーシスとクラスリン非依存性エンドサイトーシスを評価した.特に,PI3Kが関与するという報告のあるクラスリン非依存性エンドサイトーシス能を38),その指標となる蛍光標識デキストランの細胞への取り込み量で評価したところ,コントロールの細胞と比べてPI3Kγノックアウトマウス由来の細胞およびPI3K阻害薬(LY294002)で処理した細胞において取り込みが減少した.PI3KγノックアウトMEFに野生型ヒトPI3Kγを発現させるとデキストランの取り込みが回復したが,Rasと結合できない変異型PI3Kγ39)の発現(PI3Kのキナーゼ活性は保たれている)は,デキストランの取り込みを回復させなかった.これらの結果から,Ras-PI3Kシグナルがクラスリン非依存性エンドサイトーシスに関与することが示された.一方,クラスリン依存性エンドサイトーシスを蛍光標識トランスフェリンの細胞への取り込み量で評価したところ,いずれのサンプルでも変化は認められなかった.以上から,Ras-PI3Kシグナルがクラスリン非依存性エンドサイトーシスによる物質取り込みに選択的に関与することが明らかとなった.次に我々は,Ras-PI3Kシグナルが取り込みに関与する外来因子として,エンドサイトーシスで細胞に取り込まれるウイルス,特にインフルエンザウイルスに注目してその感染過程におけるRas-PI3Kシグナルの役割についての検討を行った.
4. Ras-PI3Kシグナルとインフルエンザウイルス
1)インフルエンザウイルス
インフルエンザウイルスは毎年冬季に季節的流行を繰り返し,ときに世界的流行(パンデミック)を引き起こす.直近の例では2009年に北中米を中心に発生したH1N1によるパンデミックが記憶に新しく,対策が急務とされる感染症の一つである.インフルエンザウイルスはエンベロープを持つマイナス鎖の一本鎖RNAウイルスであり,A型,B型,C型に分類される.A型インフルエンザウイルスは8本のゲノムRNAを有しており,ヘマグルチニン(hemagglutinin: HA),ノイラミニダーゼ(neuraminidase: NA),マトリクスタンパク質(M1, M2),RNAポリメラーゼ(PA, PB1, PB2),核タンパク質(nucleoprotein: NP),非構造タンパク質(nonstructural protein: NS)から構成されている.
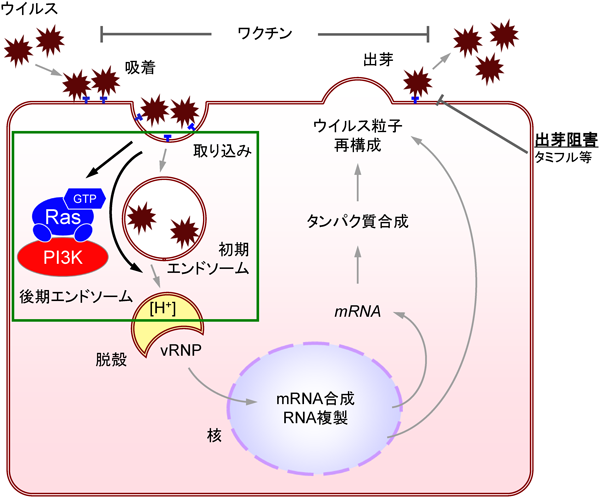
感染様式としては,図5に示すようにウイルス粒子が自身のHAと宿主細胞表面のシアル化タンパク質との結合を介して宿主細胞に吸着し,エンドサイトーシスによって宿主細胞の中に取り込まれる.
細胞内に取り込まれたウイルス粒子は,初期エンドソーム,後期エンドソームへと移行し,後期エンドソームにおけるpH低下によってHAとエンドソーム膜の融合が起こり,自身のRNAを宿主細胞に放出する.核内においてNP-PB1-PB2が複合体を形成し,ウイルスRNAが複製され,小胞体–ゴルジ装置でタンパク質が合成されるとともにウイルス粒子が再構成される.再構成されたウイルス粒子は再び細胞膜上のシアル化受容体に結合するが,NAによってその結合を切断されることで細胞外へ放出される40).
これら一連の過程を阻害する薬剤が開発され,臨床応用が検討されている.最も普及しているタミフルやリレンザはNA阻害薬であり,複製の最終過程である細胞外への放出を阻害する.しかし,一度細胞内に侵入してしまったウイルス由来のRNAには核内で複製される際に非常に高頻度に変異が導入されるため,複製以降の過程を標的とする治療法では耐性ウイルスの出現が問題となる.上記2薬剤も例外ではない41).ワクチンはウイルスに対する抗体を産生することで体内に侵入したウイルスの増殖を防いでいると考えられるが,この場合も変異による抗原性の変化の危険性に加えて,ウイルスの型が異なると効果が期待できないという問題点を有している.
2)PI3Kとインフルエンザウイルス
耐性を生じないという観点からすると,治療標的としての宿主側因子の同定が重要となるが,一般的なウイルスの研究ではウイルス側に着目した研究に比し宿主側因子の研究は遅れをとっていた.インフルエンザウイルスに関しても同様であったが,2000年代中ごろからPI3Kの感染への関与が立て続けに報告され始めた.Stephan Ludwig博士らによってPI3Kがインフルエンザウイルス感染に応答して活性化すること,およびPI3K阻害薬,wortmanninにより感染が抑制されることが報告された42,43).しかし,PI3Kの活性制御機構はウイルス由来NS1タンパク質がPI3Kの活性調整サブユニットp85βと結合し,PI3Kが活性化することが報告されたのみで44,45),その詳細はわかっていない.一方,Rasに関しては,N-Rasの過剰発現によって二本鎖RNA依存性プロテインキナーゼの活性が抑制され,免疫系が不活性化されるために,インフルエンザウイルスの感染が亢進するという報告があるのみで46),インフルエンザウイルス感染との関連はほとんどわかっていない.
3)インフルエンザウイルスの細胞内侵入過程
インフルエンザウイルスはエンドサイトーシスによって宿主細胞の中に取り込まれる.古くは電子顕微鏡による観察等から,クラスリン依存性エンドサイトーシスを介して取り込まれるという説が一般的であった47).しかし,近年ではクラスリン依存性エンドサイトーシスを阻害してもウイルス感染が成立することから48),クラスリン依存性エンドサイトーシス以外の侵入経路の存在が示唆されていた.最近,我々のグループを含めた複数のグループから,クラスリン非依存性エンドサイトーシスを介した細胞侵入が証明され,インフルエンザウイルスは複数の経路をリダンダントに利用し,細胞に侵入することが明らかになりつつある48–50).
4)Ras-PI3Kシグナルによるウイルス感染の制御
Ras-PI3Kシグナルがインフルエンザウイルス感染を制御するか検討するために,PI3KγノックアウトMEFを活用した上述の系により,インフルエンザウイルスの細胞内への取り込みとRas-PI3Kシグナルの関連を評価した.初期および後期エンドソームをマーカータンパク質であるRab5およびRab729)と蛍光タンパク質で可視化した上で,取り込まれたウイルス粒子をウイルスの核タンパク質NPに対する抗体で検出した.PI3Kγをノックアウトした細胞およびPI3Kを阻害した細胞において,ウイルスの感染が抑制された.また,デキストラン取り込みの実験と同様に,PI3Kγノックアウトマウス由来の細胞において野生型PI3Kγの発現により,ウイルス感染能が回復した一方で,Rasと結合できない変異型PI3Kγを発現させても,ウイルス感染能は回復しなかった.以上から,PI3Kがウイルス粒子の取り込みに関与すること,特に,Rasによって活性化されるPI3Kがウイルス粒子の取り込みに重要であることが示された.また,ウイルスの力価を調べる古典的な手法であるプラークアッセイを用いてウイルスの感染価を評価しても同様の結果が得られたことから,Ras-PI3Kシグナルを介した細胞内への取り込みは,感染成立にも重要であることが示唆された.
さらに,インフルエンザウイルスが細胞に取り込まれる際にRas-PI3Kシグナルが活性化されることが蛍光イメージングを用いた実験からも,生化学的な実験からも明らかとなった.以上の結果から,インフルエンザウイルスは細胞に侵入する際にRas-PI3Kシグナルを活性化し,エンドサイトーシスを亢進させることで,自らが感染しやすい環境を作り出すという巧みな手段を有していることが明らかとなった(図5).しかし,PI3Kがどのようにエンドサイトーシスとウイルスの取り込みを制御しているかは明らかではない.PI3Kの下流因子の中で最も有名なAktはウイルス感染依存的に活性化されるが,そのタイムコースはRasの活性化のそれとは異なっていた.また,Aktをノックダウン,もしくはドミナントネガティブ型のAktを細胞に発現させてもウイルス感染は抑制されなかいことからも42),Aktは細胞への取り込みには関与しないことが示唆される.
5. カルシウムシグナルを介したインフルエンザウイルスの宿主細胞取り込み機構
1)カルシウムとインフルエンザウイルス感染
これまで,インフルエンザウイルスの宿主応答因子の網羅的探索により,複数のカルシウム関連因子がウイルス感染に応答する候補として同定されている51).特に,転写・翻訳に関する因子が多く,カルシウムシグナルはウイルス粒子の複製過程に重要であるらしい51).また,高病原性鳥インフルエンザウイルス感染により,細胞外カルシウムの流入が生じ,宿主細胞がアポトーシスを生じるという報告がある52).ほかにも,インフルエンザウイルス感染により好中球の細胞内カルシウム動態が変化することや53),HAタンパク質がエンドソーム膜と融合する際にカルシウムが関与するとの報告があり54),カルシウムとインフルエンザウイルスの関連は注目を集めている.
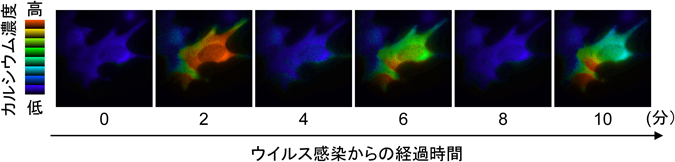
筆者らは,インフルエンザウイルスが宿主細胞のRas-PI3Kシグナルの活性化により効率的に細胞に取り込まれることを見いだしたので,その分子機構を検索した.Rasの活性化機構にはチロシンリン酸化を介する経路と,カルシウムシグナルを介する経路が知られているが55),ウイルス感染前後で宿主側タンパク質のチロシンリン酸化レベルに変化は認められなかった.一方で,カルシウム濃度変化をモニターできるFRET(Förster resonance energy transfer)センサーである“Cameleon56)”を培養細胞に発現させ,ウイルス感染時のカルシウム動態を生きた細胞で評価したところ,感染直後から複数回にわたる細胞内カルシウム濃度の一過的上昇が確認された(図6).さらに,カルシウムをキレートすると,ウイルス感染依存的なRasの活性化が阻害されたことから,ウイルスは細胞内カルシウム濃度の上昇を介してRasを活性化することが明らかとなった.なおこのカルシウム依存性のRas活性化は,カルシウム依存的なRasGEFであるCalDAG-GEF57–59)が担っているようだ.
2)インフルエンザウイルス粒子取り込みのキーレギュレータとしてのカルシウム
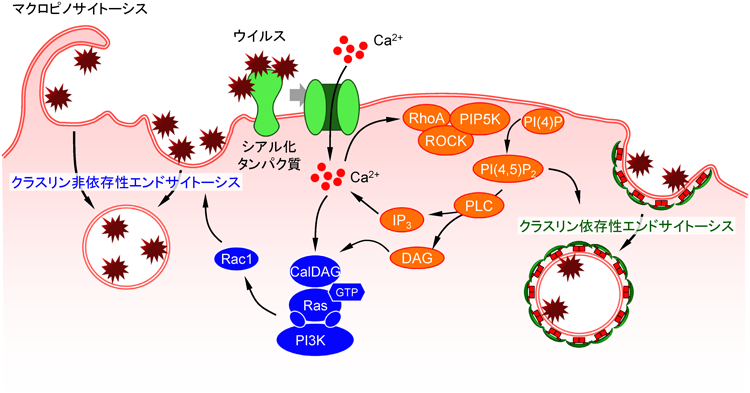
細胞内のカルシウムをキレートすると,期待どおりウイルス感染および侵入が著しく抑制された.しかし予想外に,その抑制効果はPI3K阻害時よりも強力であった.したがって,カルシウムはRas-PI3Kシグナルを介した侵入経路以外のシグナル経路も制御すると推察された.前述のとおり,インフルエンザウイルスはクラスリン依存性エンドサイトーシスおよび非依存性エンドサイトーシスの両方の経路を介して細胞に侵入することがわかっている48–50,60).そのため,細胞内カルシウムはRas-PI3K依存性のクラスリン非依存性エンドサイトーシスとクラスリン依存性エンドサイトーシスの双方を制御しているのではないかと推測した.実際,クラスリン依存性と非依存性エンドサイトーシスの単独阻害では,どちらの場合もウイルス侵入および感染はほとんど抑制されなかったが,両方のエンドサイトーシスを同時に阻害すると劇的な抑制効果が得られた.つまり,クラスリン非依存性エンドサイトーシスを抑制してもクラスリン依存性エンドサイトーシスを介して侵入し,クラスリン依存性エンドサイトーシスを抑制してもクラスリン非依存性エンドサイトーシスを介して細胞に侵入することが示唆された.カルシウムをキレートした場合は両方のエンドサイトーシスを阻害した場合と同程度の抑制効果が得られ,また,細胞内カルシウムのキレートは,クラスリン依存性および非依存性エンドサイトーシスのどちらも阻害したことから,ウイルス感染依存的な細胞内カルシウム濃度の上昇がRas-PI3Kシグナルを介したクラスリン非依存性エンドサイトーシスの制御だけでなく,クラスリン依存性エンドサイトーシスの制御にも関与すると考えられた.
実際に,膵臓β細胞や神経細胞において,カルシウムがクラスリン非依存性エンドサイトーシスを促進することが報告されている61,62).一方でシナプスにおいてはカルシウムがクラスリン非依存性エンドサイトーシスだけでなく,クラスリン依存性エンドサイトーシスに関与する事例も報告されている63).したがって,インフルエンザウイルスは,細胞が本来備えている機構をうまく活用して,エンドサイトーシスを亢進させていることが考えられる.
3)RhoAシグナルのインフルエンザウイルス感染への関与
次に,カルシウム濃度の一過的な上昇がクラスリン依存性および非依存性エンドサイトーシスを制御する機構を検討した.RhoファミリーGTP結合タンパク質はファゴサイトーシスの際のアクチン重合に関与する等の報告があり,膜輸送への関与が知られている.また,カルシウム依存的なRhoGEFも複数報告されている.以上から,カルシウムとエンドサイトーシスの両者に関与する可能性を有する候補因子として,RhoファミリーGTP結合タンパク質が浮上した.Rhoファミリーは主にRho,Rac,Cdc42からなる細胞骨格の制御因子であり,細胞運動,細胞接着,細胞周期,細胞質分裂,細胞極性,転写制御などにおいて中心的な役割を果たすことが知られている64,65).RhoA,Rac1,Cdc42のドミナントネガティブ変異体を発現した培養細胞において,ウイルス侵入と感染を評価したところ,RhoAとRac1のドミナントネガティブ変異体を発現した細胞において抑制効果が認められた.したがって,RhoAとRac1がウイルス侵入と感染に関与することが示唆された.ウイルス感染依存的なRhoAの活性化は細胞の辺縁部において一過性に生じることがFRETセンサーであるRaichu-RhoAを用いて検出することができ66),この活性化はカルシウムのキレートによって阻害された.また,Rac1のウイルス感染依存的な活性化がカルシウムのキレートとRhoAのドミナントネガティブ変異体発現によって阻害されたことから,カルシウムの下流でRhoAが,さらにその下流でRac1が働くことがわかった.一方,RhoAの活性化を担う上流因子はいまだに同定されていない.70個以上存在するRhoAのGEFのうち67),カルシウム依存的なRhoA GEFとして,PDZ-RhoGEF(ARHGEF11)が知られている68).しかし,このGEFをカルシウム依存的に活性化するチロシンキナーゼPyk2はインフルエンザウイルスとの関連が報告されておらず,PDZ-RhoGEFがインフルエンザウイルス感染に関与するGEFであるかどうかは定かではない.したがって,この分子機序の解明は今後の検討課題である.
4)複雑かつ精密なインフルエンザウイルス取り込み制御シグナル
我々はこの後,予想外の結果に直面した.RhoAがカルシウムの下流因子であることを確認するために,ウイルス感染依存的なカルシウムオシレーションがドミナントネガティブ型RhoAによって阻害されないというデータを出そうと実験したところ,RhoAのドミナントネガティブ変異体を発現させた細胞ではウイルス感染依存的なカルシウムオシレーションがまったく起こらなかったのである.さらに,野生型のRhoAはウイルス感染非依存的にカルシウムオシレーションを引き起こし,ウイルス感染依存的なオシレーションを増強した.これらのことから,RhoAはカルシウムの下流のみならず,上流因子でもあることが明らかになった.すなわち,細胞内カルシウム濃度上昇により活性化したRhoAによって,再度カルシウム濃度が上昇するという循環したシグナルが発動していることが示唆された.
幹細胞において生じているカルシウムオシレーションがRhoAによって制御されているという報告や69),血小板由来成長因子(platelet-derived growth factor: PDGF)依存的なカルシウム濃度上昇がRhoAの阻害により抑制されるという報告があり70),ウイルスは生体に備わっているシグナルネットワークをうまく活用していることが示唆される.
現在のところ,RhoAの下流シグナルは以下のところまでわかっている.ホスファチジルイノシトール-4-リン酸5-キナーゼ(phosphatidylinositol-4-phosphate 5-kinase: PIP5K)71,72)がRhoA依存的に活性化し,産生されたPIP2によりクラスリン依存性エンドサイトーシスを惹起する72).さらに,ホスホリパーゼC(phospholipase C: PLC)がPIP2をジアシルグリセロール(diacylglycerol: DAG)とIP3に分解し73),DAGとカルシウムがCalDAG-GEFに作用することで,Ras-PI3Kシグナルを介したクラスリン非依存性エンドサイトーシスを惹起する.一方,PLCによって産生されたIP3はIP3受容体に結合し,小胞体からのカルシウム放出を促し,再びRhoAを活性化することで,上記のシグナルを再び回すと考えられた.すなわち,インフルエンザウイルスはカルシウム濃度一過的上昇→RhoA活性化→カルシウム濃度一過的上昇という循環したカルシウムシグナルを発動させることで,その下流のクラスリン依存性エンドサイトーシスと非依存性エンドサイトーシスを亢進させ,細胞内に侵入しやすい環境を作り出しているというモデルが提唱された(図7).
2000年ごろからシグナル伝達の時空間制御という概念のもと,Ras自身の時空間制御の研究が盛んに行われ,その後標的因子を活性化する際の時空間的制御機構が徐々に明らかになってきた.筆者らは蛍光バイオイメージングを用いてRas-PI3Kシグナルがエンドソームで活性化されること,エンドソームから発信されるRas-PI3Kシグナルがエンドサイトーシスと,インフルエンザウイルスをはじめとする外来因子の取り込みを促進することを見いだした.今後も,生命現象の基本であるシグナル伝達とそのダイナミックな時空間制御を蛍光バイオイメージングにより解き明かし,得られた知見をもとに生命の仕組みを包括的に理解する研究はますます盛んになることと思う.
また,筆者らはインフルエンザウイルスが細胞内カルシウム濃度上昇を引き金に,Ras-PI3Kシグナルをはじめとする細胞内シグナルネットワークの活性化を介したエンドサイトーシスの亢進により細胞に取り込まれることを見いだした.現在,インフルエンザウイルスが細胞内カルシウム濃度を上昇させる機構の解明を目指している.シアル酸分解酵素処理により,ウイルス依存的なカルシウム濃度上昇が阻害されたことから,シアル化された受容体がこの機構の鍵となることが考えられ,その同定を急いでいる.インフルエンザウイルスが細胞に取り込まれる機構の全容解明をなしとげたい.
謝辞Acknowledgments
本稿は北海道大学大学院医学研究科病態医科学分野→分子細胞病理学分野→腫瘍病理学分野→細胞生理学分野と所属(名)が点々と変わる中行われた筆者らの研究成果を中心にまとめたものです.温かいご指導と叱咤激励を賜った諸先輩,共同研究や有意義なディスカッションをしていただいた多くの方々,そしていつも(研究の)世の辛酸を嘗めつつも励まし合い,ともに戦ってきた(いる)研究室の仲間たちに感謝いたします.
引用文献References
1) Murphy, J.E., Padilla, B.E., Hasdemir, B., Cottrell, G.S., & Bunnett, N.W. (2009) Proc. Natl. Acad. Sci. USA, 106, 17615–17622.
2) Grimes, M.L., Zhou, J., Beattie, E.C., Yuen, E.C., Hall, D.E., Valletta, J.S., Topp, K.S., LaVail, J.H., Bunnett, N.W., & Mobley, W.C. (1996) J. Neurosci., 16, 7950–7964.
3) Honda, K., Ohba, Y., Yanai, H., Negishi, H., Mizutani, T., Takaoka, A., Taya, C., & Taniguchi, T. (2005) Nat. Cell Biol., 434, 1035–1040.
4) Sorkin, A. & von Zastrow, M. (2009) Nat. Rev. Mol. Cell Biol., 10, 1–14.
5) Bos, J.L. (1997) Biochim. Biophys. Acta, 1333, M19–M31.
6) Downward, J. (1992) Curr. Opin. Genet. Dev., 2, 13–18.
7) Voktek, A.B. & Der, C.J. (1998) J. Biol. Chem., 273, 19925–19928.
8) Campbell, S.L., Khosravi-Far, R., Rossman, K.L., Clark, G.J., & Der, C.J. (1998) Oncogene, 17(11 Reviews), 1395–1413.
9) Cullen, P.J. & Lockyer, P.J. (2002) Nat. Rev. Mol. Cell Biol., 3, 339–348.
10) Mochizuki, N., Yamashita, S., Kurokawa, K., Ohba, Y., Nagai, T., Miyawaki, A., & Matsuda, M. (2001) Nature, 411, 1065–1068.
11) Ohba, Y., Kurokawa, K., & Matsuda, M. (2003) EMBO J., 22, 859–869.
12) York, R.D., Molliver, D.C., Grewal, S.S., Stenberg, P.E., McCleskey, E.W., & Stork, P.J. (2000) Mol. Cell. Biol., 20, 8069–8083.
13) Choy, E., Chiu, V.K., Silletti, J., Feoktistov, M., Morimoto, T., Michaelson, D., Ivanov, I.E., & Philips, M.R. (1999) Cell, 98, 69–80.
14) Chiu, V.K., Bivona, T., Hach, A., Sajous, J.B., Silletti, J., Wiener, H., Johnson, R.L. 2nd, Cox, A.D., & Philips, M.R. (2002) Nat. Cell Biol., 4, 343–350.
15) Bivona, T.G., Perez De Castro, I., Ahearn, I.M., Grana, T.M., Chiu, V.K., Lockyer, P.J., Cullen, P.J., Pellicer, A., Cox, A.D., & Philips, M.R. (2003) Nature, 424, 694–698.
16) Jura, N., Scotto-Lavino, E., Sobczyk, A., & Bar-Sagi, D. (2006) Mol. Cell, 21, 679–687.
17) Herrmann, C., Martin, G.A., & Wittinghofer, A. (1995) J. Biol. Chem., 270, 2901–2905.
18) Lemmon, M.A. (2008) Nat. Rev. Mol. Cell Biol., 9, 99–111.
19) Vanhaesebroeck, B., Guillermet-Guibert, J., Graupera, M., & Bilanges, B. (2010) Nat. Rev. Mol. Cell Biol., 11, 1–13.
20) Penuel, E. & Martin, G.S. (1999) Mol. Biol. Cell, 10, 1693–1703.
21) Gupta, S., Ramjaun, A.R., Haiko, P., Wang, Y., Warne, P.H., Nicke, B., Nye, E., Stamp, G., Alitalo, K., & Downward, J. (2007) Cell, 129, 957–968.
22) Hu, C.-D., Chinenov, Y., & Kerppola, T.K. (2002) Mol. Cell, 9, 789–798.
23) Hu, C.-D. & Kerppola, T.K. (2003) Nat. Biotechnol., 21, 539–545.
24) Tsutsumi, K., Fujioka, Y., Tsuda, M., Kawaguchi, H., & Ohba, Y. (2009) Cell. Signal., 21, 1672–1679.
25) Gruenberg, J. & van der Goot, F.G. (2006) Nat. Rev. Mol. Cell Biol., 7, 495–504.
26) Christoforidis, S., Miaczynska, M., Ashman, K., Wilm, M., Zhao, L., Yip, S.C., Waterfield, M.D., Backer, J.M., & Zerial, M. (1999) Nat. Cell Biol., 4, 249–252.
27) Bohdanowicz, M., Cosio, G., Backer, J.M., & Grinstein, S. (2010) J. Cell Biol., 191, 999–1012.
28) Doherty, G.J. & McMahon, H.T. (2009) Annu. Rev. Biochem., 78, 857–902.
29) Huotari, J. & Helenius, A. (2011) EMBO J., 30, 3481–3500.
30) Parton, R.G. & del Pozo, M.A. (2013) Nat. Rev. Mol. Cell Biol., 14, 98–112.
31) Mayor, S. & Pagano, R.E. (2007) Nat. Rev. Mol. Cell Biol., 8, 603–612.
32) Rupper, A., Lee, K., Knecht, D., & Cardelli, J. (2001) Mol. Biol. Cell, 12, 2813–2824.
33) Posor, Y., Eichhorn-Gruenig, M., Puchkov, D., Schöneberg, J., Ullrich, A., Lampe, A., Müller, R., Zarbakhsh, S., Gulluni, F., Hirsch, E., Krauss, M., Schultz, C., Schmoranzer, J., Noé, F., & Haucke, V. (2013) Nature, 499, 1–7.
34) Li, G., D’Souza-Schorey, C., Barbieri, M.A., Cooper, J.A., & Stahl, P.D. (1997) J. Biol. Chem., 272, 10337–10340.
35) Tall, G.G., Barbieri, M.A., Stahl, P.D., & Horazdovsky, B.F. (2001) Dev. Cell, 1, 73–82.
36) Porat-Shliom, N., Kloog, Y., & Donaldson, J.G. (2008) Mol. Biol. Cell, 19, 765–775.
37) Sasaki, T., Irie-Sasaki, J., Jones, R.G., Oliveira-dos-Santos, A.J., Stanford, W.L., Bolon, B., Wakeham, A., Itie, A., Bouchard, D., Kozieradzki, I., Joza, N., Mak, T.W., Ohashi, P.S., Suzuki, A., & Penninger, J.M. (2000) Science, 287, 1040–1046.
38) Araki, N., Johnson, M.T., & Swanson, J.A. (1996) J. Cell Biol., 135, 1249–1260.
39) Pacold, M.E., Suire, S., Perisic, O., Lara-Gonzalez, S., Davis, C.T., Walker, E.H., Hawkins, P.T., Stephens, L., Eccleston, J.F., & Williams, R.L. (2000) Cell, 103, 931–943.
40) Watanabe, T., Watanabe, S., & Kawaoka, Y. (2010) Cell Host Microbe, 7, 427–439.
41) Samson, M.L., Pizzorno, A., Abed, Y., & Boivin, G. (2013) Antiviral Res., 98, 174–185.
42) Ehrhardt, C., Marjuki, H., Wolff, T., Nurnberg, B., Planz, O., Pleschka, S., & Ludwig, S. (2006) Cell. Microbiol., 8, 1336–1348.
43) Ehrhardt, C. & Ludwig, S. (2009) Cell. Microbiol., 11, 863–871.
44) Hale, B.G., Jackson, D., Chen, Y.-H., Lamb, R.A., & Randall, R.E. (2006) Proc. Natl. Acad. Sci. USA, 103, 14194–14199.
45) Li, Y., Anderson, D.H., Liu, Q., & Zhou, Y. (2008) J. Biol. Chem., 283, 23397–23409.
46) Bergmann, M., Romirer, I., Sachet, M., Fleischhacker, R., Garcia-Sastre, A., Palese, P., Wolff, K., Pehamberger, H., Jakesz, R., & Muster, T. (2001) Cancer Res., 61, 8188–8193.
47) Doxsey, S.J., Brodsky, F.M., Blank, G.S., & Helenius, A. (1987) Cell, 50, 453–463.
48) Sieczkarski, S.B. & Whittaker, G.R. (2002) J. Virol., 76, 10455–10464.
49) Fujioka, Y., Tsuda, M., Hattori, T., Sasaki, J., Sasaki, T., Miyazaki, T., & Ohba, Y. (2011) PLoS ONE, 6, e16324.
50) de Vries, E., Tscherne, D.M., Wienholts, M.J., Cobos-Jiménez, V., Scholte, F., García-Sastre, A., Rottier, P.J.M., & de Haan, C.A.M. (2011) PLoS Pathog., 7, e1001329.
51) König, R., Stertz, S., Zhou, Y., Inoue, A., Hoffmann, H.H., Bhattacharyya, S., Alamares, J.G., Tscherne, D.M., Ortigoza, M.B., Liang, Y., Gao, Q., Andrews, S.E., Bandyopadhyay, S., De Jesus, P., Tu, B.P., Pache, L., Shih, C., Orth, A., Bonamy, G., Miraglia, L., Ideker, T., García-Sastre, A., Young, J.A.T., Palese, P., Shaw, M.L., & Chanda, S.K. (2009) Nature, 463, 813–817.
52) Ueda, M., Daidoji, T., Du, A., Yang, C.S., Ibrahim, M.S., Ikuta, K., & Nakaya, T. (2010) J. Virol., 84, 3068–3078.
53) Hartshorn, K., Collamer, M., Auerbach, M., Myers, J., Pavlotsky, N., & Tauber, A. (1988) J. Immunol., 141, 1295–1301.
54) Zhukovsky, M.A., Markovic, I., & Bailey, A.L. (2007) Arch. Biochem. Biophys., 465, 101–108.
55) Vigil, D., Cherfils, J., Rossman, K.L., & Der, C.J. (2010) Nat. Rev. Cancer, 10, 842–857.
56) Nagai, T., Yamada, S., Tominaga, T., Ichikawa, M., & Miyawaki, A. (2004) Proc. Natl. Acad. Sci. USA, 101, 10554–10559.
57) Stone, J.C., Ebinu, J.O., Bottorff, D.A., Chan, E.Y.W., Stang, S.L., & Dunn, R.J. (1998) Science, 280, 1082–1086.
58) Kawasaki, H., Springett, G.M., Toki, S., Canales, J.J., Harlan, P., Blumenstiel, J.P., Chen, E.J., Bany, I.A., Mochizuki, N., Ashbacher, A., Matsuda, M., Housman, D.E., & Graybiel, A.M. (1999) Proc. Natl. Acad. Sci. USA, 96, 318–318.
59) Yamashita, S., Mochizuki, N., Ohba, Y., Tobiume, M., Okada, Y., Sawa, H., Nagashima, K., & Matsuda, M. (2000) J. Biol. Chem., 275, 25488–25493.
60) Chen, C. & Zhuang, X. (2008) Proc. Natl. Acad. Sci. USA, 105, 11790–11795.
61) He, Z., Fan, J., Kang, L., Lu, J., Xue, Y., Xu, P., Xu, T., & Chen, L. (2008) Traffic, 9, 910–923.
62) Kabayama, H., Nakamura, T., Takeuchi, M., Iwasaki, H., Taniguchi, M., Tokushige, N., & Mikoshiba, K. (2009) Mol. Cell. Neurosci., 40, 27–38.
63) Wu, X.-S., McNeil, B.D., Xu, J., Fan, J., Xue, L., Melicoff, E., Adachi, R., Bai, L., & Wu, L.-G. (2009) Nat. Neurosci., 12, 1003–1010.
64) Etienne-Manneville, S. & Hall, A. (2002) Nature, 420, 629–635.
65) Norman, K.R., Fazzio, R.T., Mellem, J.E., Espelt, M.V., Strange, K., Beckerle, M.C., & Maricq, A.V. (2005) Cell, 123, 119–132.
66) Yoshizaki, H., Ohba, Y., Kurokawa, K., Itoh, R.E., Nakamura, T., Mochizuki, N., Nagashima, K., & Matsuda, M. (2003) J. Cell Biol., 162, 223–232.
67) Cook, D.R., Rossman, K.L., & Der, C.J. (2014) Oncogene, 33, 4021–4035.
68) Ying, Z.K., Giachini, F.R.C., Tostes, R.C., & Webb, R.C. (2009) Arterioscler. Thromb. Vasc. Biol., 29, 1657–1663.
69) Kim, T.-J., Seong, J., Ouyang, M., Sun, J., Lu, S., Hong, J.P., Wang, N., & Wang, Y. (2009) J. Cell. Physiol., 218, 285–293.
70) Chong, L.D., Traynor-Kaplan, A., Bokoch, G.M., & Schwartz, M.A. (1994) Cell, 79, 507–513.
71) De Lange, G., Birk, M., Boersma, D., Dercksen, J., Dmitriev, P., Ermakov, A.B., Filippenko, L.V., Golstein, H., Hoogeveen, R.W.M., de Jong, L., Khudchenko, A.V., Kinev, N.V., Kiselev, O.S., van Kuik, B., de Lange, A., van Rantwijk, J., Selig, A.M., Sobolev, A.S., Torgashin, M.Y., de Vries, E., Wagner, G., Yagoubov, P.A., & Koshelets, V.P. (2002) J. Cell. Biochem., 226, 10455–10464.
72) Roth, M.G., Padron, D., Wang, Y.J., Yamamoto, M., & Yin, H. (2003) J. Cell Biol., 162, 693–701.
73) Horowitz, L.F., Hirdes, W., Suh, B.-C., Hilgemann, D.W., Mackie, K., & Hille, B. (2005) J. Gen. Physiol., 126, 243–262.
著者寸描
 藤岡 容一朗(ふじおか よういちろう)
藤岡 容一朗(ふじおか よういちろう)
北海道大学大学院医学研究科特任助教.博士(学術).
略歴
1979年東京都に生る.2003年東北大学工学部卒業.08年東京大学大学院総合文化博士課程修了.08年より北海道大学大学院医学研究科にて博士研究員,11年より学振特別研究員(PD).14年より現職.
研究テーマと抱負
生きた細胞においてシグナル伝達を可視化しています.面白いと自分で思えるのはもちろん,他の研究者だけでなく一般の方にも面白いと思ってもらえるような研究をしていきたいです.
ウェブサイト
http://cp.med.hokudai.ac.jp
趣味
サッカー.
 大場 雄介(おおば ゆうすけ)
大場 雄介(おおば ゆうすけ)
北海道大学大学院医学研究科教授.博士(医学).
略歴
1970年北海道に生る.96年北海道大学医学部卒業.2000年同大学院医学研究科修了.国際医療センター研究所(流動研究員),阪大微研(助手),東大院医(助手),北大院医(准教授)を経て12年より現職.01~05年JSTさきがけ研究員(兼任).
研究テーマと抱負
細胞内シグナル伝達をイメージングで可視化し,細胞機能発現のメカニズムや生命のしくみを明らかにしていきたい.また,自身が行った研究によって1ページ,いや1行でもいいので,教科書を書き換えるような成果を出したい.
ウェブサイト
http://cp.med.hokudai.ac.jp
趣味
音楽鑑賞,スポーツ観戦,ドライブ.