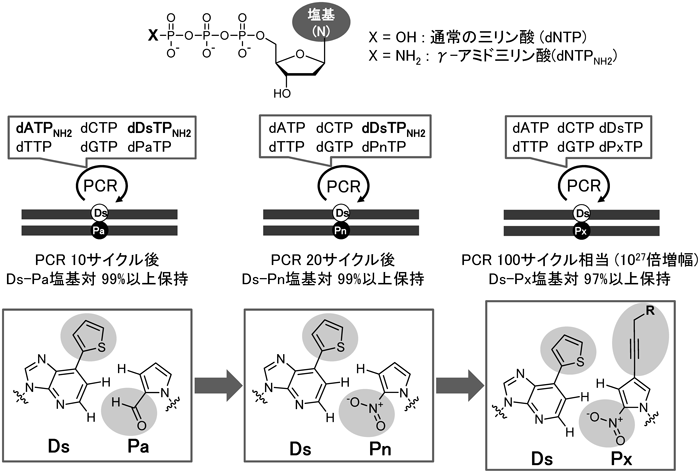
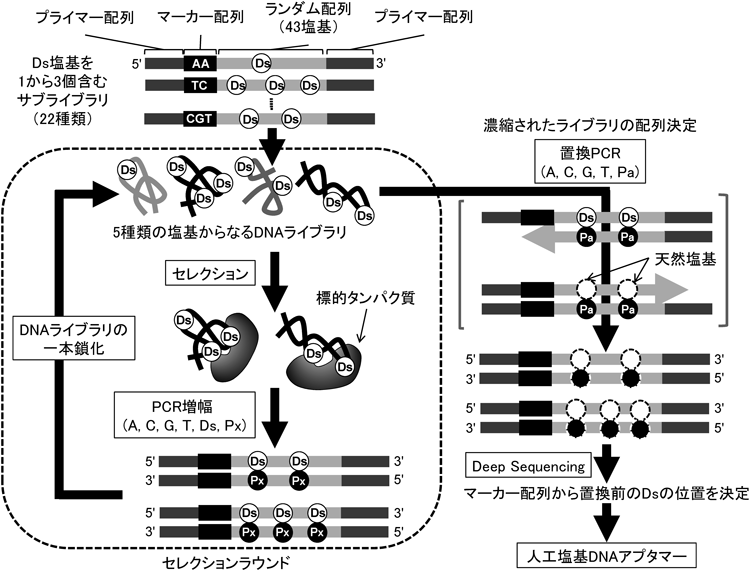
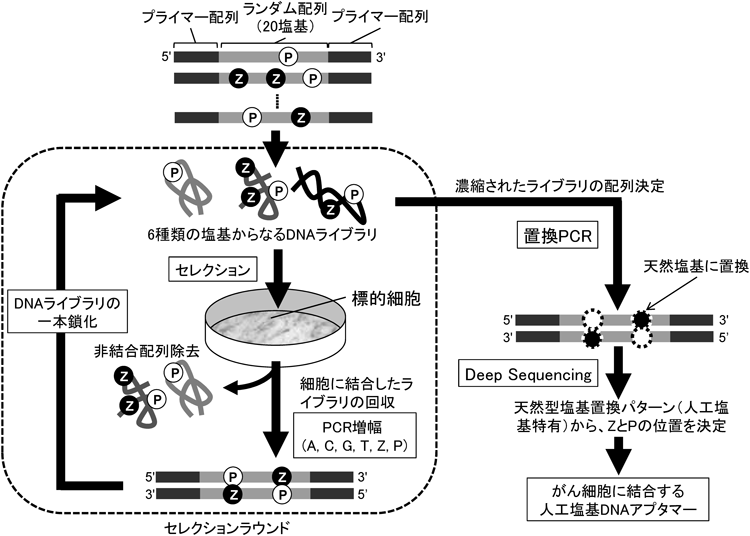
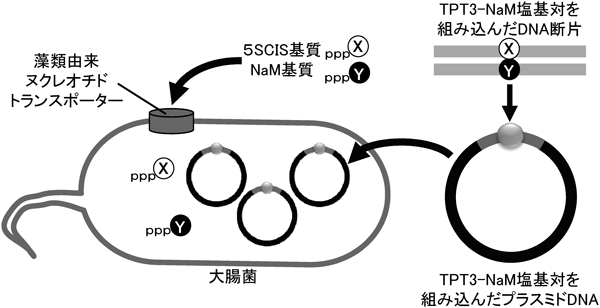
21) Hirao, I., Ohtsuki, T., Fujiwara, T., Mitsui, T., Yokogawa, T., Okuni, T., Nakayama, H., Takio, K., Yabuki, T., Kigawa, T., Kodama, K., Yokogawa, T., Nishikawa, K., & Yokoyama, S. (2002) Nat. Biotechnol., 20, 177–182.
57) Gold, L., Ayers, D., Bertino, J., Bock, C., Bock, A., Brody, E.N., Carter, J., Dalby, A.B., Eaton, B.E., Fitzwater, T., Flather, D., Forbes, A., Foreman, T., Fowler, C., Gawande, B., Goss, M., Gunn, M., Gupta, S., Halladay, D., Heil, J., Heilig, J., Hicke, B., Husar, G., Janjic, N., Jarvis, T., Jennings, S., Katilius, E., Keeney, T.R., Kim, N., Koch, T.H., Kraemer, S., Kroiss, L., Le, N., Levine, D., Lindsey, W., Lollo, B., Mayfield, W., Mehan, M., Mehler, R., Nelson, S.K., Nelson, M., Nieuwlandt, D., Nikrad, M., Ochsner, U., Ostroff, R.M., Otis, M., Parker, T., Pietrasiewicz, S., Resnicow, D.I., Rohloff, J., Sanders, G., Sattin, S., Schneider, D., Singer, B., Stanton, M., Sterkel, A., Stewart, A., Stratford, S., Vaught, J.D., Vrkljan, M., Walker, J.J., Watrobka, M., Waugh, S., Weiss, A., Wilcox, S.K., Wolfson, A., Wolk, S.K., Zhang, C., & Zichi, D. (2010) PLoS ONE, 5, e15004.
59) Sefah, K., Yang, Z., Bradley, K.M., Hoshika, S., Jimenez, E., Zhang, L., Zhu, G., Shanker, S., Yu, F., Turek, D., Tan, W., & Benner, S.A. (2014) Proc. Natl. Acad. Sci. USA, 111, 1449–1454.