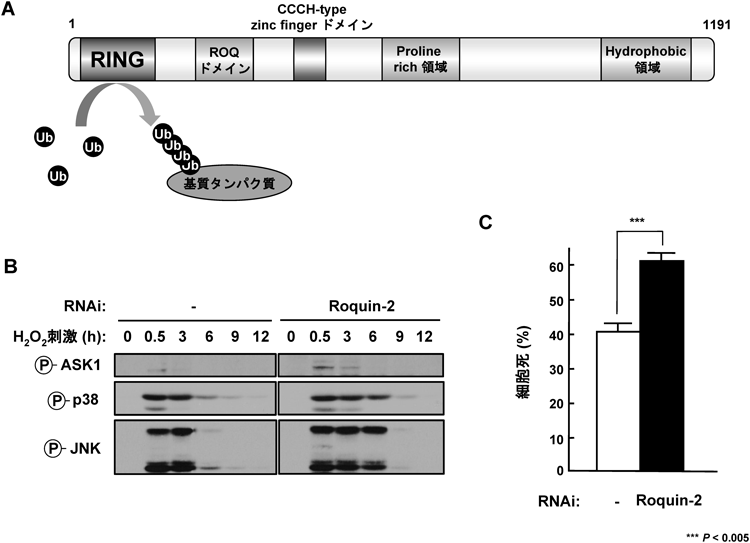
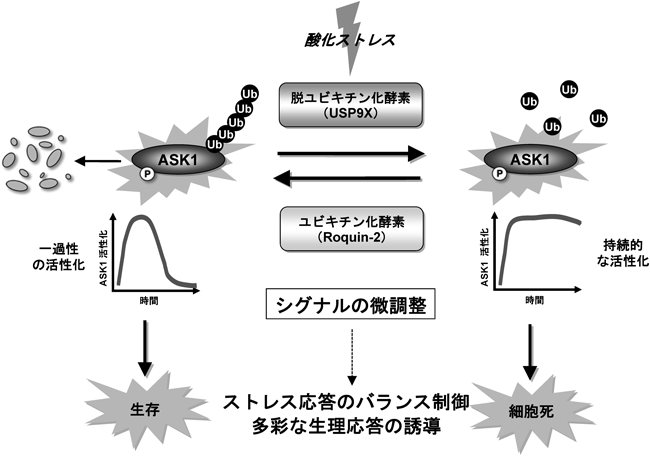
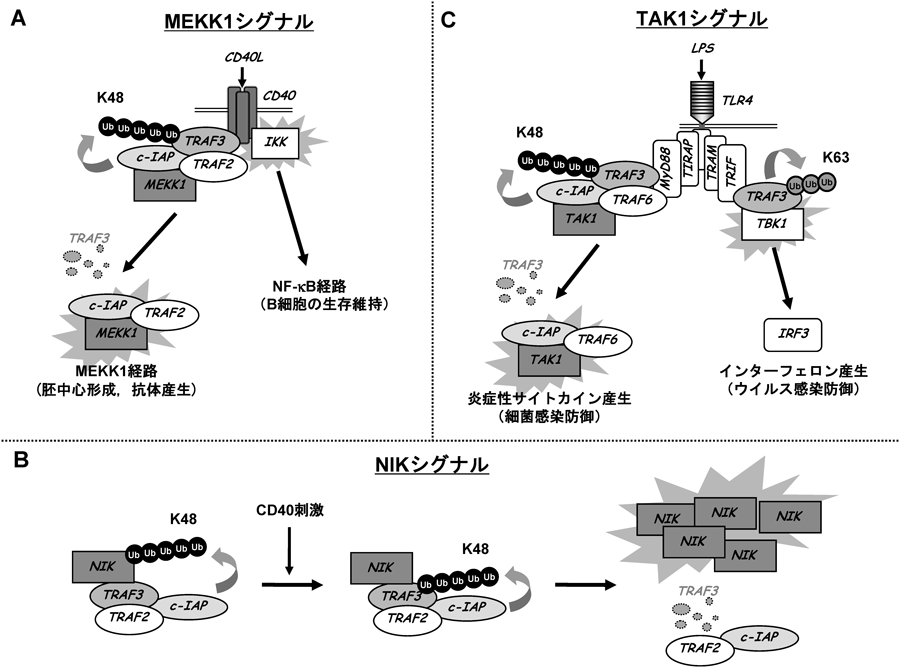
多様なユビキチン化酵素群によるリン酸化シグナルの制御機構Regulatory mechanisms of phosphorylation signaling by various ubiquitin-related enzymes
東北大学大学院薬学研究科衛生化学分野Laboratory of Health Chemistry, Graduate School of Pharmaceutical Sciences, Tohoku University ◇ 〒980-8578 宮城県仙台市青葉区荒巻字青葉6-36-3 Aoba, Aramaki, Aoba-ku, Sendai-shi, Miyagi 980-8578, Japan
発行日:2015年2月25日Published: February 25, 2015