生命にとり自己複製は必須の属性で,このためには複製過程で遺伝情報を安定に維持しながら倍化する必要がある.真核細胞は遺伝情報を自己の核内にクロマチンの形で保持しており1),これらの情報はDNAの塩基配列に依存したゲノム情報と,それ以外(DNA修飾,ヒストン修飾)のエピゲノム情報に大別される.これらの情報の多くは細胞周期の複製期(S期)に正確に複製されて,分裂期(M期)に娘細胞に正確に分配される2,3).これらの機構は,常に各プロセスと情報状態を認識する機構により監視されており,いったん異常が認知されると細胞はプロセスの進行を停止し,適切に情報を修復する機能を持っている.細胞周期が正確にG1期(ギャップ1),S期,G2期(ギャップ2),M期を一方向性に進行するのもこの監視機構によると考えられている4).すなわち,正常状態では真核細胞は自己のDNA複製が完了するまで決して染色体分配を行わない.DNA複製と染色体分配の連動制御が破綻すると,ゲノム不安定状態が惹起され遺伝情報の損傷や齟齬につながる.最近,DNA複製過程におけるさまざまな内因的ストレスが,がんなどの染色体不安定疾患の主要な原因であると考えられるようになった5–7).一方,遺伝情報は常に紫外線,放射線,化学物質などの外的ストレスにさらされており,多かれ少なかれ損傷を受けると考えられている.細胞はこれら内因的,あるいは外的ストレスにより受けたゲノム損傷に対し適切に応答することで,異常な遺伝情報を持った細胞の蓄積を防いでいる.このような細胞応答が破綻すると,“がん”や“老化促進”といった増殖異常症を誘発すると考えられる8,9).一方,多細胞生物体ではゲノム情報のみならず,多様な形質を持つ個々の細胞が形質を維持しながら増殖する必要がある.このことは,ゲノム以外の遺伝情報であるエピゲノム情報もDNA複製と連動して複製される必要があることを示している10).本稿では,これまでの筆者らの研究成果を中心に,①DNA複製と染色体分配の連動機構とDNA複製起点からの複製開始,②DNA損傷に応答したゲノム情報安定維持機構,③ゲノム状態監視機構の破綻とがん化と細胞老化,④DNA複製と連動したDNAメチル化維持機構とその破綻について概説し,遺伝情報不安定化の分子基盤と疾患発症の病理的機構について議論する.
2. DNA複製と染色体分配の連動機構とDNA複製起点からの複製開始
哺乳動物細胞は細胞周期と呼ばれる一連のプロセスを通じて安定的に自己複製を行っている.これらのプロセスは不可逆的かつ一方向性であり,各プロセスの完了と次のプロセスの開始は厳密に連動している.細胞周期を安定的に,正確な順番で,一方向性の進行を担保する分子機構は,酵母からヒトに至る真核細胞で基本的に維持されていると考えられている.この機構をチェックポイント機構と呼び,多くはプロテインキナーゼのカスケード反応により制御されている11).このうち,DNA複製の完了をモニターし,染色体分配を開始する分子機構はS-Mチェックポイントと呼ばれ12),ゲノム情報を安定的に維持する主要機構であり,この破綻は大規模なゲノム不安定性を惹起すると考えられている.しかしながら,この分子機構の本体についてはほとんど理解されていなかった.
出芽酵母におけるMec1, 分裂酵母におけるRad3はほとんどの異常DNA構造をモニターするチェックポイントの中心的キナーゼで,主要な最終分子標的はM期の開始を誘導するサイクリンB-Cdk1複合体である13).ヒトを含めた哺乳動物細胞におけるMec/Rad3の相同分子はATR(ataxia telangiectasia and Rad3 related)であり,その下流で酵母同様にChk1(checkpoint kinase 1)を制御する14).したがって筆者らは,哺乳動物細胞におけるS-MチェックポイントはATRを介したシグナルによって制御されていると考えた.しかしながら,哺乳動物細胞においてATR,Chk1は生存に必須のキナーゼで,S-Mチェックポイントへの関与を解析することは困難であった15–17).そこで,Cre/loxPシステムを用いてChk1のコンディショナル欠失ES細胞を作製し解析した18).
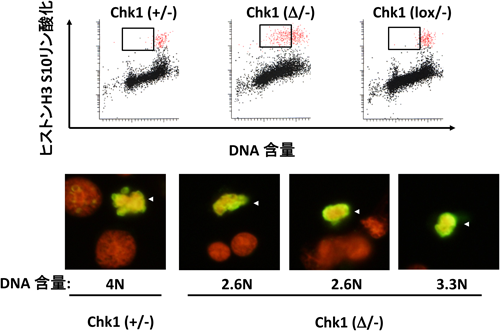
Chk1遺伝子をES細胞において欠失すると,細胞増殖が著明に抑制され細胞死が誘導されることがわかった.レーザー走査型サイトメトリー(laser scanning cytometer)を用いて解析したところ,Chk1lox/−細胞では90%以上のヒストンH3セリン10リン酸化(pH 3S10;分裂期マーカー)陽性細胞が4N DNA含量を示したが,Chk1Δ/−細胞では60%のpH 3S10陽性細胞が4N以下のDNA含量であった.さらに各細胞におけるクロマチン凝集の程度を測定したところ,Chk1lox/−細胞では90%程度のクロマチン凝集細胞が4N DNA含量を示したにも関わらず,Chk1Δ/−細胞では60%程度の凝集細胞が4N以下のDNA含量を示した(図1).このことは,Chk1がDNA複製完了をモニターし,染色体分配を制御するS-Mチェックポイントに必須の役割を果たしていることを示している.ごく最近コンディショナルATR欠損DT40細胞を用いた解析から同様にATRがS-Mチェックポイントに重要な役割を果たしていることが報告された19).実際,正常のS期にATR依存的にChk1がリン酸化されることがわかった.Chk1リン酸化特異的抗体を用いた解析から,セリン317はS期を通してリン酸化されているが,セリン345はS期後期に特異的にリン酸化されていた20).したがって,これらのChk1リン酸化がS-Mチェックポイントにおいて重要な役割を果たしていると考えられた.また,Chk1−/−細胞においてはCdk1分子のチロシン15のリン酸化が著しく低下していることも明らかとなり,Chk1によるS-Mチェックポイントの標的はサイクリンB-Cdk1複合体であることがわかった.すなわち,S期においてはATR依存的リン酸化Chk1によりCdk1チロシン15のリン酸化が誘導されることでM期の開始が抑制されており,DNA複製が完了するとATR不活性化とともにChk1が脱リン酸化され,サイクリンB-Cdk1が活性化されてM期が開始すると考えられる.
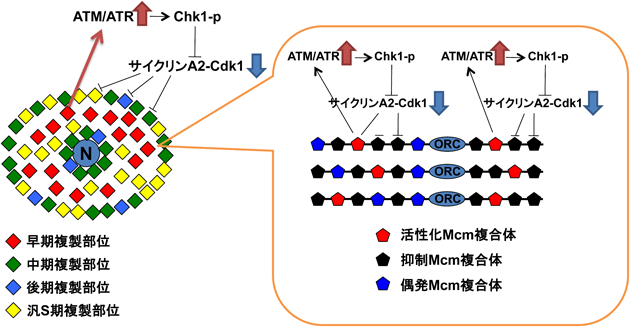
興味深いことに,DNA複製期におけるATR依存的Chk1リン酸化は,サイクリンB-Cdk1を介したM期開始のみならず,後期複製起点からの複製開始も制御することがわかった21).真核細胞では複製起点からの複製開始がS期を通じて時空的に制御されていることが知られている.哺乳動物細胞の複製レプリコンは潜在的に数百から数千存在していると考えられているが,これら複製レプリコンは同時に複製を開始するわけではない.活性化したレプリコンはクラスターを形成しており,ブロモデオキシウリジン染色によりfociとして観察される22).さらに,一つのレプリコンには複数のMcm2~7複合体が存在しているが,そのうちごく少数のMcm複合体が選択されて複製が開始する23).すなわち,哺乳動物細胞の複製起点は,Mcm複合体の選択と,活性化レプリコンクラスター形成という二つのレベルで時空的に制御されている.興味深いことに,ヒトにおけるDNA変異率はS期後期に複製される領域に高いことが報告され24),DNA複製起点の制御はゲノム情報安定化維持に重要な役割を果たしていると考えられる.Chk1−/−細胞を用いた解析から,Chk1非存在下では異常なDNA複製起点からの複製開始,すなわち後期複製起点からの複製がS期早期に開始することがわかった21).さらに,DNA複製起点からの複製開始が亢進した結果,複製フォークの進行が著しく遅くなることもわかった.同様の結果はATR欠損細胞でも確認された25).このChk1によるDNA複製起点制御の標的はサイクリンA2-Cdk1複合体であることもわかった(図2).出芽酵母においてもClb5依存的なCdk活性が特異的に後期複製起点からの複製開始を制御していること26)と合わせると,DNA複製起点からの複製制御は酵母からヒトまで保存されているのかもしれない.
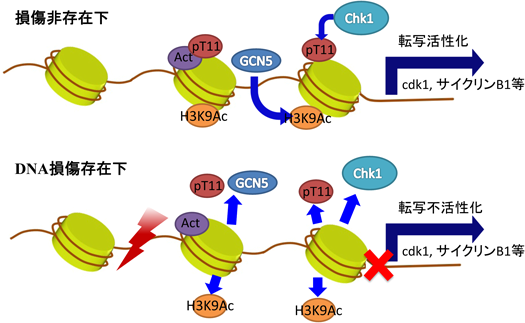
細胞はDNA損傷を受けると自己のゲノム情報を修復・維持するためにさまざまな細胞応答を示す.DNA損傷に応じた細胞周期停止や,DNA修復機構の活性化などについては比較的理解が進んでいたが,DNA損傷に応じた転写制御については不明な点が多かった.DNA損傷によりさまざまな遺伝子の発現が変化することが知られている.たとえば,細胞に紫外線を照射すると発現量が3倍以上変化する遺伝子は全遺伝子の4%にもおよび,このうちの90%は転写抑制される遺伝子である27).DNA損傷を受けた後に転写が活性化する遺伝子については,p53やNF-κBといったストレス応答転写因子の解析から多くの知見が得られているが,転写抑制機構はほとんど理解されていなかった.ヒストンのN末端側(ヒストンテール)は,アセチル化,メチル化,リン酸化,ユビキチン化などのさまざまな化学修飾を受け,クロマチンの多様な機能を生み出している28).筆者らは転写制御に重要な役割を果たすヒストン修飾がDNA損傷後どのように変化するか解析した.ヒストンH3分子のリシン9(K9),ヒストンH2A分子のリシン5(K5),ヒストンH4分子のリシン8(K8)のアセチル化の減少とともに,ヒストンH3のトレオニン11(T11)のリン酸化が急激に低下することを見いだした29).ヒストンH3のセリン10(S10)のリン酸化は,M期における染色体凝集や,間期においてはK9メチル化に影響して転写を制御している30).したがって,T11のリン酸化も同様に転写制御に関与している可能性がある.実際,T11のリン酸化はS10リン酸化同様にM期において著しく亢進していたが,間期においても認められた.これまでT11のリン酸化はDlk1により触媒されることが報告されたが,DNA損傷に応じたT11のリン酸化変化にDlk1が関与するかどうかはまったく不明であった31).筆者らはDNA損傷に応じたT11リン酸化変化に関与するキナーゼの同定を試みたところ,Chk1キナーゼがT11のリン酸化を触媒することがわかった.重要なことに,Chk1はDNA損傷非存在下ではクロマチンに結合しているが,DNA損傷後にATR依存的なリン酸化によりクロマチンから遊離する20,32).したがって,T11のリン酸化の減少はChk1のクロマチンからの遊離と一致しており,Chk1がDNA損傷非存在下で働くT11リン酸化酵素と考えて矛盾がないと思われた.実際,Chk1のATR依存的リン酸化部位変異体(S317A/S345A)を細胞内に導入すると,DNA損傷後にもChk1変異体はクロマチンから遊離せず,T11のリン酸化減少や転写の抑制も阻害された.T11のリン酸化減少が転写抑制を引き起こす分子基盤について解析したところ,T11がリン酸化されたヒストンH3 N末端ペプチドは,ヒストンH3K9をアセチル化する酵素であるGCN5との相互作用が,非リン酸化ペプチドと比較して約20倍強いことが明らかとなった.DNA損傷後に転写が低下するサイクリンB1あるいはCdk1遺伝子のプロモーター領域では,DNA損傷後のChk1の解離とT11リン酸化の低下,およびGCN5の解離とK9アセチル化の低下が認められた.これらの結果から筆者らは,DNA損傷非存在化においてChk1はサイクリンB1あるいはCdk1遺伝子プロモーター上のクロマチンに結合しており,ヒストンH3T11をリン酸化してGCN5のプロモーター結合を促進すると考えた.プロモーター上のGCN5はヒストンH3K9をアセチル化することによりこれら遺伝子の転写を活性化している.DNAに損傷が生じると活性化したATR依存的にChk1がリン酸化を受けてプロモーター上のクロマチンから解離する.Chk1の解離に伴いT11の脱リン酸化が誘導されGCN5のプロモーター結合が抑制される.GCN5のプロモーターからの解離はK9の脱アセチル化を引き起こし,最終的に転写抑制が誘導されると考えられる(図3).T11を介した同様の転写の抑制が,アンドロゲン依存的な転写制御に重要な役割を果たしていることが報告された33).このときT11をリン酸化する酵素はPRK1であることも明らかとなった.ごく最近,SET1/MLLサブユニットの一つであるWDR5がT11リン酸化を認識して結合し,MLL複合体をプロモーター領域に集積することでヒストンH3K4のトリメチル化を亢進することで転写を活性化することが示された34).
それではDNA損傷に応答したT11の脱リン酸化を触媒するホスファターゼは何であろうか? ホスファターゼ阻害剤であるオカダ酸を用いた実験から,PP1(protein phosphatase 1)ファミリーのホスファターゼがこの脱リン酸化を触媒すると考えられた.遺伝子ノックダウン法を用いてさらに解析を進めると,PP1γが特異的にDNA損傷に応答したT11の脱リン酸化を触媒していることがわかった35).実際,PP1γ活性はDNA損傷後にCdk依存的なT311のリン酸化抑制により増強した.細胞からPP1γを欠失させるとDNA損傷に対して感受性が亢進することもわかった.これらの結果は,T11のリン酸化がATRにより大きく2経路を介して制御されていることを示している.すなわち,ATRの活性化はChk1リン酸化を介してプロモーターからのChk1の解離を促進し,一方Chk1依存的なCdk活性の抑制はPP1γ分子のT311の脱リン酸化を誘導してホスファターゼ活性を増強することから協調してT11の脱リン酸化を制御していると考えられる.
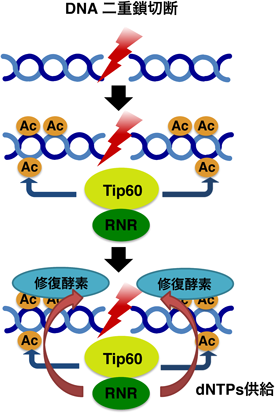
DNA損傷が起こった場合,DNA損傷の種類に依存して細胞は適切に,かつ特異的にDNA損傷修復経路を活性化させ,損傷DNAを修復する.ほとんどのDNA損傷修復においては新たなDNA合成を必要とするが,DNA合成にあたっては適切な濃度のdNTPsがDNA鎖新生のために供給されなければならない.DNA合成過程での不適切な濃度のdNTPs供給は高い変異率をもたらし,ゲノム情報の不安定化を引き起こす.dNTPs合成の律速酵素はリボヌクレオチド還元酵素(ribonucleotide reductase: RNR)で,大サブユニットR1と小サブユニットR2のヘテロ四量体から構成される36).出芽酵母においては,細胞内dNTPsの量的制御はチェックポイント機構により制御されている.一つはRad53-DUN1経路により転写抑制複合体がリン酸化を受けて転写抑制機能が阻害されて,RNR遺伝子の発現が亢進する37).またDUN1はRNRの阻害サブユニットであるSml1をリン酸化して分解することでRNR活性を増強する38).さらに,出芽酵母のRNRはWtm1/Dif1依存的にその細胞内局在が変化し,dNTPs供給を制御している39).筆者らは同様の機構が哺乳動物細胞にも保存されていると考え,DNA損傷に応答したdNTPs供給機構について解析を行った.興味深いことに,哺乳動物細胞では酵母と異なり,DNA損傷時には細胞全体のdNTPs量はほとんど変化しないことが報告されている40).哺乳動物1細胞の容量は0.5 pL程度と考えられているため,S期以外の時期において細胞内dNTPs濃度は0.5 µM程度となる41).DNA複製あるいは修復に機能するDNAポリメラーゼの多くは,10 µM程度のKm値を示すためDNA損傷部位に限局したdNTPsの供給機構が正確なDNA修復に必要となると考えられる.筆者らは,哺乳動物細胞においてDNA損傷後にRNRが損傷部位にすばやく集積することを見いだした42).この集積にはTip60ヒストンアセチル化酵素が必要であることもわかった.実際,Tip60はDNA損傷後に損傷部位に最も早く集積するタンパク質の一つであると考えられている43).損傷部位に集積したTip60は損傷部位周辺のヒストンH4分子のアセチル化を誘導し,さまざまな修復関連因子の集積を促進するとともに,RNRを局在化させて適切な濃度のdNTPs供給に機能していると考えられる(図4).このTip60依存的なRNRの損傷部位への集積は特にG1期でのDNA損傷修復に重要な役割を果たしており,この集積が阻害されると細胞はDNA損傷に対して高感受性を示すようになる.ごく最近,IRBIT(IP3R binding protein released with inositol 1,4,5-trisphosphate)がdATP依存的にRNRに結合し,酵素活性を阻害することが報告された44).これらの知見は,哺乳動物細胞においても酵母同様に,RNRが細胞内局在,発現調節,およびRNR阻害タンパク質により重層的に制御されていることを意味しており,dNTPs濃度の適切な調整はゲノム情報の安定維持に必須の役割を果たしていると考えられる.
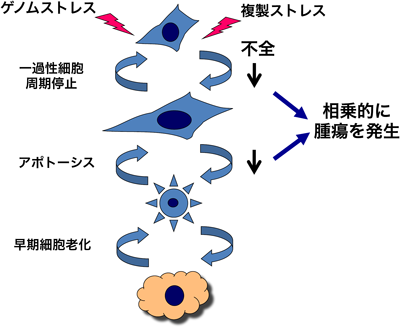
細胞周期の進行不全や,ゲノム状態の不全はDNA損傷応答機構を活性化する.DNA損傷応答機構は,一過性細胞周期停止やアポトーシス,あるいは早期細胞老化を誘導して,ゲノム情報の修復や異常ゲノム情報を持った細胞の自己複製を強く抑制する.これらの機構は個体内で抗腫瘍機構として機能していると考えられている.したがって,DNA損傷応答シグナルを発信する細胞周期進行における各プロセスの完了や,ゲノム状態を監視する分子機構の不全は容易に“がん”などの染色体不安定症を引き起こすと予想される.しかしながら,ATM(ataxia telangiectasia mutated)あるいはATRの下流で情報を下流分子に伝達するチェックポイントキナーゼ,Chk1およびChk2の変異マウスは発がん率の亢進を示さない(Chk1の場合はホモ欠失により胎生致死となるため,ヘテロマウスの解析結果である)16,17,45).これらの結果は,Chk1およびChk2により伝達されるシグナルが“ゲノム安定性に関わっているもののがん発症には関連しない”,あるいはこれら経路が“がん発症に関して相補的な機能を果たしている”のどちらかを示唆しているものと考えられる.これらを明らかにする目的で,筆者らはChk1+/−Chk2−/−二重変異マウスの発がん率,およびマウス由来の胎仔線維芽細胞のゲノム安定維持状態について解析を行った46).興味深いことに,それぞれ単独の変異マウスのがん発症率は長期間の飼育後も亢進しなかったが,Chk1+/−Chk2−/−二重変異マウスは遅発型の高自然発がん率を示した.胎仔線維芽細胞の解析から,Chk1+/−由来の細胞ではDNA損傷に応じたG2期停止能が著しく低下しており,この低下はChk2欠失によりさらなる影響を受けなかった.一方,Chk2−/−由来の細胞ではDNA損傷に応じたp53依存的なアポトーシス誘導が損なわれていた.このアポトーシス誘導不全はChk1片アリルの欠損でまったく影響を受けなかった.これらの結果は,一過性細胞周期停止あるいはアポトーシス誘導のどちらかが機能すれば個体において抗腫瘍効果として十分機能することを意味しており,両方が機能不全に陥るとゲノム不安定性が惹起されて“がん”発症に至ると考えられる(図5).実際,Chk1+/−Chk2−/−二重変異細胞では異常染色体が高率に存在しており,またストレス非存在化においてもp53の蓄積やγH2AXシグナルを認めた.しかしながら,早期細胞老化が個体内においていかなるレベルで抗腫瘍効果を示すかについては不明のままであった.
細胞老化は不可逆的増殖停止を主徴とする細胞表現型で,今から50年ほど前にHayflickらによりヒトの正常細胞がある一定の増殖回数の後に恒久的な増殖停止を示すことから見いだされた47).細胞老化研究が進むにつれて,Rasなどのがん遺伝子の活性化や酸化的ストレス,あるいはDNA損傷によっても細胞老化と同様の増殖停止が誘導されることも明らかとなり,がん防御機構の一つとして重要な役割を果たしていることが提唱された48).さらに,動脈硬化,糖尿病などの老化関連疾患や生活習慣病にも関与していることが明らかになりつつある49,50).細胞老化誘導機構についてはこれまで多くの研究成果が報告されており,がん抑制遺伝子産物p53やpRbが必須の役割を果たしていることがわかっているが51),具体的にこれら産物がどのように機能することで細胞老化が誘導されるのかまったく不明であった.
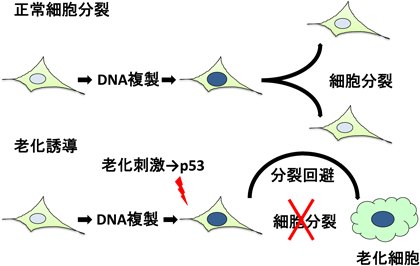
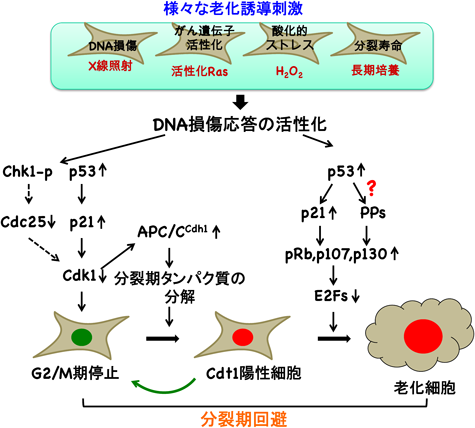
筆者らは老化誘導機構の全貌を明らかにする目的で,まず老化細胞がどの細胞周期相で細胞周期から逸脱し,増殖を停止するのかを理研の宮脇らにより開発されたFUCCIシステムを用いて解析した52).このシステムではG1期の細胞が赤色に,S期からG2期の細胞が緑色に蛍光発色することから生細胞において細胞周期相を同定することが可能となる.したがって,この系を用いれば一生細胞の老化過程をタイムラプス撮影で観察することが可能となる.その結果,非常に興味深いことに分裂寿命,がん遺伝子活性化,DNA損傷などのすべての細胞老化誘導刺激において,大部分の正常線維芽細胞は細胞分裂期に入ることなく,緑色の細胞(SCF優位の細胞)が赤(APC/C優位の細胞)に変化することがわかった53).この現象が,真に細胞が分裂期を回避してG1期に移行していることを示しているのかどうかを明らかにする目的で,DNA損傷後の分裂期を経ずに赤く変化した細胞にSV40 largeT抗原を発現させたところ,DNA損傷後24時間の細胞は分裂期に移行したのに対して,DNA損傷後48時間の細胞ではDNA再複製が誘導され八倍体細胞が生じた.同様に活性型サイクリンB1-Cdk1融合タンパク質を導入したところ,24時間までの細胞は分裂期に移行したのに対して,48時間後の細胞では分裂期への誘導はみられなかった.重要なことにDNA損傷後24時間から48時間の間に細胞分裂期の進行に必要となるサイクリンB1やCdk1,あるいはCdc20という分裂期タンパク質がほぼ完全に消失することが示された.これらの結果は,分裂期を経ずに赤く変化した細胞が実際にG1期に移行していることを示しており,正常線維芽細胞は老化刺激が加わると分裂期を回避してG1期に移行することがわかった(図6).またこの分裂期回避は,分裂期に必要となる分裂期タンパク質のG2期における消失により誘導されることも示唆された.これらのことから,老化細胞はG1期4倍体細胞であることがわかった.
それではこのG2期タンパク質の消失による分裂期回避がどのような機構で制御されているのか,また老化誘導に必要不可欠であるのかを解析した.がん抑制遺伝子産物p53は細胞老化誘導に必須であることがわかっているので,p53を欠失した線維芽細胞でFUCCIシステムを用いて同様に解析した.その結果,p53欠失細胞は老化誘導刺激後分裂期に移行するが,異常な細胞分裂を示し最終的に二つの細胞に分裂した.したがって,老化誘導刺激による細胞分裂期回避にp53は必須の役割を果たしている.興味深いことに,G2期細胞でp53を一過性に発現誘導するのみで細胞分裂回避が起こり,細胞老化が誘導されることがわかった.この細胞老化誘導はp53をG2期以外の時期に発現させてもまったく起こらなかった.このことは,p53依存的分裂期回避が細胞老化誘導に必要不可欠な役割を果たしていることを示している.
p53はどのようにして分裂期回避を誘導しているのであろうか? p53の代表的な下流分子であるp21をG2期の細胞に一過性に発現させると,FUCCIの解析で分裂期を経ることなく緑色の細胞から赤色細胞への変化は起こるが,G1期への移行や細胞老化誘導は起こらず,p53の下流のp21以外の経路も分裂期回避に関わっていることが示唆された.しかしながら,p21の発現誘導はAPC/CCdh1のG2期での早期活性化を誘導し,分裂期タンパク質の分解を促進していることがわかった.一方,老化誘導刺激後,分裂期タンパク質遺伝子の転写はp53依存的に強く抑制され,この抑制はp53の下流でRbファミリータンパク質(pRb, p107, p130)に依存していることがわかった.これらの結果から,分裂期回避を誘導する分裂期タンパク質の消失は,p53の下流でのp21-APC/CCdh1の早期活性化による分解の促進と,Rbファミリータンパク質による転写抑制の二つの経路により制御されていることを示している(図7).重要なことに,細胞周期のG2期で活性型のCdh1とRbを一過性に発現させただけで分裂期が回避され,その結果細胞老化が誘導されることがわかった.予想されたとおり,これら二つのタンパク質をG2期以外の時期に発現させても細胞老化は誘導されなかった.一方,これまで細胞老化誘導に重要と考えられていたp16は細胞老化状態の維持に必須の役割を果たしていたが,老化誘導にはまったく影響を与えなかった.
それではこの分裂期回避は個体レベルでの細胞老化誘導にどのような役割を果たしているのだろうか? 母斑細胞性母斑(ほくろ)はBRAF遺伝子に変異を持つ色素細胞の良性腫瘍で,老化細胞のマーカーの一つであるSA-β-Gal(老化関連β-ガラクトシダーゼ)陽性であることから,個体におけるがん遺伝子活性化誘導老化細胞と考えられている54).そこで,母斑細胞のDNA含量と細胞周期について解析を行った.興味深いことに,母斑細胞は同じ組織切片の表皮細胞,あるいは血管内皮細胞と比較してほぼ2倍のDNA含量を示し,サイクリンB1陰性,およびKi67陰性であることから,G1期四倍体細胞であることがわかった.以上の結果は,個体内においても分裂期回避は細胞老化誘導に重要な役割を果たしており,培養細胞でみられた老化誘導機構は個体内においても機能していることを強く示唆している.今回筆者らの研究により老化誘導機構の一端が明らかになったことにより,老化防止法の開発や,新たながん治療法,あるいは老化関連病予防法の確立に結びつくことが期待される.
5. DNA複製と連動したDNAメチル化維持機構とその破綻
多細胞生物体は個々の細胞のもつ多様な形質をエピゲノム情報により規定している.したがって,固有の細胞形質を維持しながら増殖するためには細胞周期の過程でゲノム情報のみならず,エピゲノム情報も正確に複製され娘細胞に分配されなければならない.DNAメチル化は最も安定なエピゲノム情報の一つであり,多くはゲノム配列上のCpG配列のシトシン残基の5位の炭素にメチル基が付加されることにより生じる.DNAメチル化はメチルDNA結合タンパク質を集積することでクロマチン構造変換を誘導し転写抑制に機能していると考えられている55).分化した細胞は固有のDNAメチル化パターンを持っており,分化細胞が増殖する際に正確に娘細胞に伝達される.しかしながら,古典的DNA複製装置ではDNAメチル化は複製されないため,DNA複製後に一過性に片鎖DNAのみメチル化されたヘミメチル化DNAが生じる.ヘミメチル化DNAは維持DNAメチル化酵素であるDnmt1により触媒されて両鎖ともメチル化されたフルメチル化DNAに変換される.この変換のためにはDnmt1がDNA複製部位,すなわちヘミメチル化部位に集積する必要があるが,ヘミメチル化DNA結合タンパク質Uhrf1が必要なこと以外ほとんどその分子機構についてはわかっていなかった56–59).Uhrf1はユビキチン様ドメインや,タンデムTUDOR-PHDドメイン,SRAドメイン,RINGフィンガードメインを持つ多機能タンパク質である.
筆者らは,DNA維持メチル化機構を明らかにする目的で,アフリカツメガエル卵細胞抽出液を用いた生化学的解析を行った.アフリカツメガエル卵間期抽出液にツメガエル精子核を加えると,試験管内でDNA複製と共役したクロマチン形成を再現することができる60).まずこの系を用いてDNA維持メチル化が試験管内で再構成できるかどうか解析した.卵間期抽出液に精子核とともにメチル化の供与体としてメチル基に放射性ラベルしたS-アデノシルメチオニンを加えてDNAへのメチル基の取り込みを解析した.その結果,DNA複製と共役し,Uhrf1に依存したDNAメチル化が観察された61).さらに,DNA複製と一致してDnmt1がクロマチンに結合すること,またこの結合はUhrf1に依存していることがわかった.これらの結果は,DNA維持メチル化機構がアフリカツメガエル卵抽出液を用いた解析で再構成可能であることを示している.
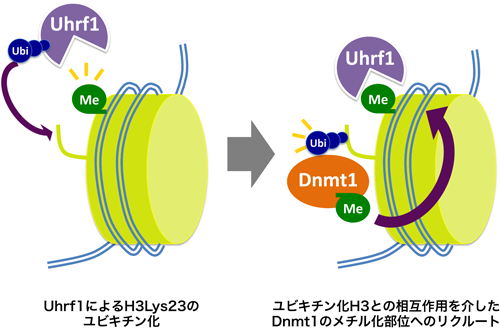
興味深いことに,Dnmt1を卵抽出液から抗体を用いて除去するとUhrf1がクロマチン上に過剰蓄積することがわかった.さらにこのとき,Uhrf1の蓄積と一致してヒストンH3がユビキチン化されることがわかった.このユビキチン化部位を質量分析により解析したところ,ヒストンH3分子のリシン23が特異的なユビキチン化標的であることもわかった.それではこのユビキチン化はDNA維持メチル化にどのような役割を果たしているのだろうか? 筆者らはこのヒストンH3ユビキチン化がヘミメチル化部位へのDnmt1集積に機能していると考えて,ユビキチン化ヒストンH3に結合するタンパク質を探索した.その結果,Dnmt1が直接ユビキチン化ヒストンH3に結合することが免疫沈降,あるいはFar-westernブロット法により明らかとなった.これまで,Dnmt1がDNA複製部位に集積するためにはDnmt1分子のreplication foci targeting sequence(RFTS)領域が必要であることが報告されている62).実際,RFTS領域を欠損させたDnmt1はユビキチン化ヒストンH3とは結合しないことも示された.以上の結果から,Uhrf1依存的なヒストンH3ユビキチン化が直接結合を介してDnmt1をDNA複製部位に集積するのに重要な役割を果たしていることが示唆された(図8).
重要なことに,このUhrf1依存的ヒストンH3リシン23ユビキチン化は哺乳動物細胞でもDNA複製期特異的に検出された.さらに,Uhrf1欠損細胞にユビキチン化活性を欠失した変異Uhrf1を発現させた細胞の解析から,ヒストンH3リシン23のユビキチン化が起こらないと,Dnmt1のDNA複製部位への局在が抑制され,さらにES細胞の解析からゲノム全領域やレトロトランスポゾン領域のDNAメチル化レベルが著明に減少することが明らかとなった.これらの結果は,Uhrf1によるヒストンH3ユビキチン化がDNA維持メチル化に必須の役割を果たしていることを示しており,エピゲノム修飾の安定複製機構の一端が明らかとなったと考えている.
これまで20年来,“哺乳動物細胞がどのように固有の性質を維持しながら、安定に増殖することができるのか”という非常にシンプル,かつ重要な命題を明らかにするため研究を続けてきた.以前は,ゲノム情報が安定に複製・維持される機構についてもほとんどわかっていなかったが,DNA複製機構や修復機構,あるいは細胞周期チェックポイント機構の詳細が明らかになり徐々にではあるが体系的に理解可能になってきた.エピゲノム情報について以前はその重要性についても不明な点が多かったが,今ではその役割や機能について理解が深まっている.ではあるが,エピゲノム情報の安定的複製機構についてはまさに研究の緒についたばかりであり,これからの10年,20年での大きな進歩が期待される.とりわけ,ヒストン修飾がどのように複製されるかについては現在ほとんどその実体がわかっていないが,最近の多くの最新解析技術の進歩により解明が進むであろう.またエピゲノム情報をプログラムどおりに書き換える“細胞分化や発生過程”がどのようにエピゲノム情報安定維持機構と関わっているのか非常に興味深い.一方,ゲノム・エピゲノムの安定維持機構の破綻がいかなる分子機構により,がん,老化などの病態を引き起こしていくのかについてはいまだ大きな謎が多く,治療法や予防法の確立を考えると“依然暗闇にいる”といっても過言ではないだろう.今後,ゲノム・エピゲノム情報の安定複製機構を解明することで,人類を脅かす疾患克服に貢献していきたいと考えている.
引用文献References
1) Luger, K., Dechassa, M.L., & Tremethick, D.J. (2012) Nat. Rev. Mol. Cell Biol., 13, 436–447.
2) Nurse, P. (1994) Cell, 79, 547–550.
3) Alavert, C. & Groth, A. (2012) Nat. Rev. Mol. Cell Biol., 13, 157–167.
4) Murray, A.W. (1992) Nature, 359, 599–604.
5) Bartek, J., Lukas, C., & Lukas, J. (2004) Nat. Rev. Mol. Cell Biol., 5, 792–804.
6) Lecona, E. & Fernandez-Capetillo, O. (2014) Exp. Cell Res., S0014-4827 (14) 00424-8, 329, 26–34.
7) Hills, S.A. & Diffley, J.F. (2014) Curr. Biol., 24, R435–R444.
8) Harper, J.W. & Elledge, S.J. (2007) Mol. Cell, 28, 739–745.
9) Niida, H. & Nakanishi, M. (2006) Mutagenesis, 21, 3–9.
10) Liu, Q. & Gong, Z. (2011) Curr. Opin. Plant Biol., 14, 187–194.
11) Elledge, S.J. (1996) Science, 274, 1664–1672.
12) Enoch, T., Carr, A.M., & Nurse, P. (1992) Genes Dev., 6, 2035–2046.
13) Carr, A.M. (1997) Curr. Opin. Genet. Dev., 7, 93–98.
14) Paulsen, R.D. & Cimprich, K.A. (2007) DNA Repair (Amst.), 6, 953–966.
15) Brown, E.J. & Baltimore, D. (2000) Genes Dev., 14, 397–402.
16) Takai, H., Tominaga, K., Motoyama, N., Minamishima, Y.A., Nagahama, H., Tsukiyama, T., Ikeda, K., Nakayama, K., Nakanishi, M., & Nakayama, K. (2000) Genes Dev., 14, 1439–1447.
17) Liu, Q., Guntuku, S., Cui, X.S., Matsuoka, S., Cortez, D., Tamai, K., Luo, G., Carattini-Rivera, S., DeMayo, F., Bradley, A., Donehower, L.A., & Elledge, S.J. (2000) Genes Dev., 14, 1448–1459.
18) Niida, H., Tsuge, S., Katsuno, Y., Konishi, A., Takeda, N., & Nakanishi, M. (2005) J. Biol. Chem., 280, 39246–39252.
19) Eykelenboom, J.K., Harte, E.C., Canavan, L., Pastor-Peidro, A., Calvo-Asensio, I., Llorens-Agost, M., & Lowndes, N.F. (2013) Cell Reports, 5, 1095–1107.
20) Niida, H., Katsuno, Y., Banerjee, B., Hande, M.P., & Nakanishi, M. (2007) Mol. Cell. Biol., 27, 2572–2581.
21) Katsuno, Y., Suzuki, A., Sugimura, K., Okumura, K., Zineldeen, D.H., Shimada, M., Niida, H., Mizuno, T., Hanaoka, F., & Nakanishi, M. (2009) Proc. Natl. Acad. Sci. USA, 106, 3184–3189.
22) Jackson, D.A. (1995) BioEssays, 17, 587–591.
23) Woodward, A.M., Gohler, T., Luciani, M.G., Oehlmann, M., Ge, X., Gartner, A., Jackson, D.A., & Blow, J.J. (2006) J. Cell Biol., 173, 673–683.
24) Stamatoyannopoulos, J.A., Adzhubei, I., Thurman, R.E., Kryukov, G.V., Mirkin, S.M., & Sunyaev, S.R. (2009) Nat. Genet., 41, 393–395.
25) Shechter, D., Costanzo, V., & Gautier, J. (2004) Nat. Cell Biol., 6, 648–655.
26) Donaldson, A.D., Raghuraman, M.K., Friedman, K.L., Cross, F.R., Brewer, B.J., & Fangman, W.L. (1998) Mol. Cell, 2, 173–182.
27) Gentile, M., Latonene, L., & Laiho, M. (2003) Nucleic Acids Res., 31, 4779–4790.
28) Kouzarides, T. (2007) Cell, 128, 693–705.
29) Shimada, M., Niida, H., Zineldeen, D.H., Tagami, H., Tanaka, M., Saito, H., & Nakanishi, M. (2008) Cell, 132, 221–232.
30) Cheung, P., Tanner, K.G., Cheung, W.L., Sassone-Corsi, P., Denu, J.M., & Allis, C.D. (2000) Mol. Cell, 5, 905–915.
31) Preuss, U., Landsberg, G., & Scheidtmann, K.H. (2003) Nucleic Acids Res., 31, 878–885.
32) Smits, V.A., Reaper, P.M., & Jackson, P. (2006) Curr. Biol., 16, 150–159.
33) Metzger, E., Yin, N., Wissmann, M., Kunowska, N., Fischer, K., Friedrichs, N., Patnaik, D., Higgins, J.M., Potier, N., Scheidtmann, K.H., Buettner, R., & Schule, R. (2008) Nat. Cell Biol., 10, 53–60.
34) Kim, J.Y., Banerjee, T., Vinckevicius, A., Luo, Q., Parker, J.B., Baker, M.R., Radhakrishnan, I., Wei, J.J., Barish, G.D., & Chakravarti, D. (2014) Mol. Cell, 54, 613–625.
35) Shimada, M., Haruta, M., Niida, H., Sawamoto, K., & Nakanishi, M. (2010) EMBO Rep., 11, 883–889.
36) Reichard, P. (1993) Science, 260, 1773–1777.
37) Huang, M., Zhou, Z., & Elledge, S.J. (1998) Cell, 94, 595–605.
38) Zhao, X. & Rothstein, R. (2002) Proc. Natl. Acad. Sci. USA, 99, 3746–3751.
39) Lee, Y.D., Wang, J., Stubbe, J., & Elledge, S.J. (2008) Mol. Cell, 32, 70–80.
40) Hakanson, P., Hofer, A., & Thelander, L. (2006) J. Biol. Chem., 281, 7834–7841.
41) Imaizumi, T., Jean-Louis, F., Dubertret, M.L., Bailly, C., Cicurel, L., Petchot-Bacque, J.P., & Dubertret, L. (1996) J. Dermatol. Sci., 11, 134–141.
42) Niida, H., Katsuno, Y., Sengoku, M., Shimada, M., Yukawa, M., Ikura, M., Ikura, T., Kohno, K., Shima, H., Suzuki, H., Tashiro, S., & Nakanishi, M. (2010) Genes Dev., 24, 333–338.
43) Sun, Y., Jiang, X., & Price, B.D. (2010) Cell Cycle, 9, 930–936.
44) Arnaoutov, A. & Dasso, M. (2014) Science, 345, 1512–1515.
45) Takai, H., Naka, K., Okada, Y., Watanabe, M., Harada, N., Saito, S., Anderson, C.W., Appella, E., Nakanishi, M., Suzuki, H., Nagashima, K., Sawa, H., Ikeda, K., & Motoyama, N. (2002) EMBO J., 21, 5195–5205.
46) Niida, H., Murata, K., Shimada, M., Ogawa, K., Ohta, K., Suzuki, K., Fujigaki, H., Khaw, A.K., Banerjee, B., Hande, M.P., Miyamoto, T., Miyoshi, I., Shirai, T., Motoyama, N., Delhase, M., Appella, E., & Nakanishi, M. (2010) EMBO J., 29, 3558–3570.
47) Hayflick, L. & Moorhead, P.S. (1961) Exp. Cell Res., 25, 585–621.
48) Campisi, J. & d’Adda di Fagagna, F. (2007) Nat. Rev. Mol. Cell Biol., 8, 729–740.
49) Minamino, T., Miyauchi, H., Yoshida, T., Ishida, Y., Yoshida, H., & Komuro, I. (2002) Circulation, 105, 1541–1544.
50) Minamino, T., Miyauchi, H., Tateno, K., Kunieda, T., & Komuro, I. (2004) Cell Cycle, 3, 449–451.
51) Busuttil, R.A., Dolle, M., Campisi, J., & Vijga, J. (2004) Ann. N. Y. Acad. Sci., 1019, 245–255.
52) Sakaue-Sawano, A., Kurokawa, H., Morimura, T., Hanyu, A., Hama, H., Osawa, H., Kashiwagi, S., Fukami, K., Miyata, T., Miyoshi, H., Imamura, T., Ogawa, M., Masai, H., & Miyawaki, A. (2008) Cell, 132, 487–498.
53) Johmura, Y., Shimada, M., Misaki, T., Naiki-Ito, A., Miyoshi, H., Motoyama, N., Ohtani, N., Hara, E., Nakamura, M., Morita, A., Takahashi, S., & Nakanishi, M. (2014) Mol. Cell, 55, 73–84.
54) Michaloglou, C., Vredeveld, L.C., Soengas, M.S., Denoyelle, C., Kuilman, T., van der Horst, C.M., Majoor, D.M., Shay, J.W., Mooi, W.J., & Peeper, D.S. (2005) Nature, 436, 720–724.
55) Prokhortchouk, E. & Defossez, P.A. (2008) Biochim. Biophys. Acta, 1783, 2167–2173.
56) Bostick, M., Kim, J.K., Esteve, P.O., Clark, A., Pradhan, S., & Jacobsen, S.E. (2007) Science, 317, 1760–1764.
57) Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T.A., Shinga, J., Mizutani-Koseki, Y., Toyoda, T., Okamura, K., Tajima, S., Mitsuya, K., Okano, M., & Koseki, H. (2007) Nature, 450, 908–912.
58) Arita, K., Ariyoshi, M., Tochio, H., Nakamura, Y., & Shirakawa, M. (2008) Nature, 455, 818–821.
59) Hashimoto, H., Horton, J.R., Zhang, X., Bostick, M., Jacobsen, S.E., & Cheng, X. (2008) Nature, 455, 826–829.
60) Arias, E.E. & Walter, J.C. (2004) Front. Biosci., 9, 3029–3045.
61) Nishiyama, A., Yamaguchi, L., Sharif, J., Johmura, Y., Kawamura, T., Nakanishi, K., Shimamura, S., Arita, K., Kodama, T., Ishikawa, F., Koseki, H., & Nakanishi, M. (2013) Nature, 502, 249–253.
62) Leonhardt, H., Page, A.W., Weier, H.U., & Bestor, T.H. (1992) Cell, 71, 865–873.
著者紹介Author Profile
 中西 真(なかにし まこと)
中西 真(なかにし まこと)名古屋市立大学大学院医学研究科細胞生化学分野教授.博士(医学).
略歴1960年名古屋市に生まれる.85年名古屋市立大学医学部卒業,89年同大学院博士課程修了後,自治医科大学助手,講師,国立長寿医療研究センター室長,名古屋市立大学医学部助教授を経て,2000年名古屋市立大学医学部生化学第二講座教授.02年改組により名古屋市立大学大学院医学研究科教授.現在に至る.
研究テーマと抱負最近は自分の衰えを感じながら,なぜヒトは老いるのか?ということに興味を持っています.
趣味アウトドアスポーツならなんでも.