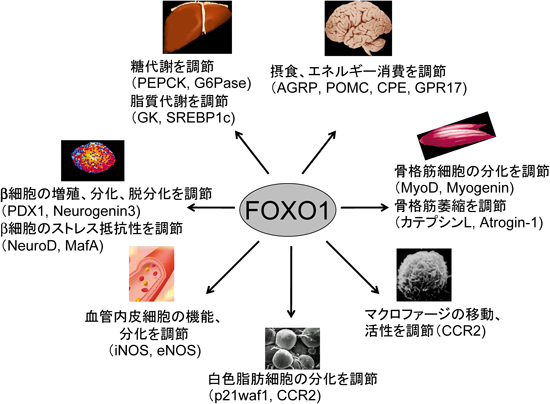
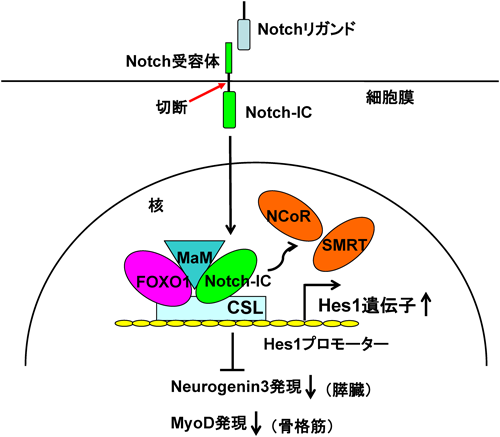
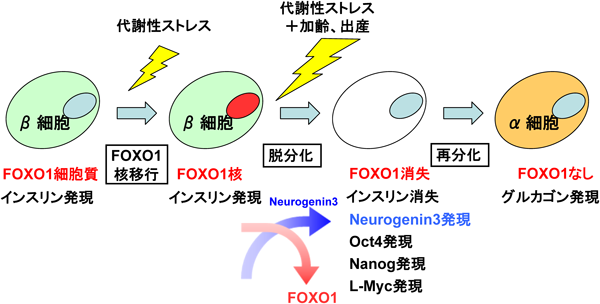
各臓器におけるFOXO1の代謝作用The roles of FOXO1 in various metabolic organs
1 群馬大学生体調節研究所代謝シグナル研究展開センターMetabolic Signal Research Center, Institute for Molecular and Cellular Regulation, Gunma University ◇ 〒371-8512 群馬県前橋市昭和町3-39-153-39-15 Showa-machi, Maebashi-shi, Gunma 371-8512, Japan
2 藤沢湘南台病院Fujisawa Shounandai Hospital ◇ 〒252-0802 神奈川県藤沢市高倉23452345 Takakura, Fujisawa-shi, Kanagawa 252-0802, Japan
発行日:2015年4月25日Published: April 25, 2015