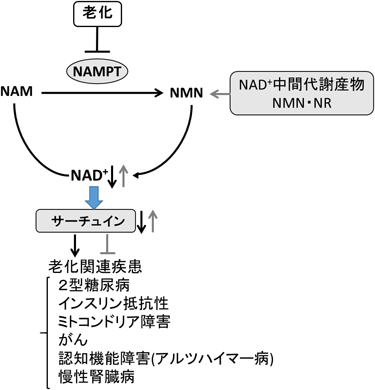
ニコチンアミドアデニンジヌクレオチド(nicotinamide adenine dinucleotide: NAD+)は,すべての生物種に存在する古典的な補酵素であり,酸化還元反応で中心的役割を果たす.近年,哺乳類NAD+合成系の鍵酵素であるニコチンアミドホスホリボシルトランスフェラーゼ(nicotinamide phosphoribosyltransferase: NAMPT)が環境・栄養状態に応答することでNAD+量を調節し,サーチュインに代表されるNAD+消費酵素を介して代謝,炎症,分化,老化などの生物学的な多彩な局面において重要な役割を果たすことが明らかにされてきた1–3).さらに,インスリン抵抗性,糖尿病,がんおよびアルツハイマー病に代表される老化関連疾患においてNAMPTの酵素反応産物であるニコチンアミドモノヌクレオチド(nicotinamide mononucleotide: NMN)や,ニコチンアミドリボシド(nicotinamide riboside: NR)などのNAD+中間代謝産物がNAD+量を増加させ,病態を改善することも報告されている(表1).こうした基礎研究成果を基盤として,NAD+合成系やその主要メディエーターとして知られるサーチュインを標的とした新しいトランスレーショナル型リサーチの機運が高まっている.本稿では,NAD+合成系の老化関連疾患における病態生理学的意義と創薬標的としての可能性を,最新の知見を交えて考察したいと思う.
表1 NAD中間代謝産物投与効果| NAD中間代謝産物 | 投与対象 | 投与量 | 投与経路 | 投与期間 | 投与効果 | 参考文献 |
|---|
| NMN | 高脂肪食負荷誘導性糖尿病マウス,老化糖尿病マウス | 500 mg/kg | 腹腔内 | 7~10日間 | NAD+合成系,SIRT1が触媒する反応速度が上昇し,耐糖能異常,インスリン抵抗性,脂質異常症を改善. | [7] |
| 老年マウス(22月齢) | 500 mg/kg | 腹腔内 | 7日間 | 加齢に伴う骨格筋のミトコンドリア機能不全を回復させ,骨格筋萎縮や炎症のマーカー,インスリンシグナル伝達,グルコース取り込みを改善. | [9] |
| 心筋虚血モデルマウス | 500 mg/kg | 腹腔内 | 心筋虚血30分前 | 心臓NAD+量を増加させ,SIRT1を介してFOXO1を脱アセチル化し虚血再灌流障害に対し保護的に働く. | [21] |
| 心筋ミトコンドリア呼吸鎖複合体Iの機能低下マウス | 500 mg/kg | 腹腔内 | 1日2回3日間 | 心筋におけるNAD+/NADH比の上昇,ミトコンドリアタンパク質の脱アセチル化を亢進し,ミトコンドリア膜透過性遷移孔の透過性を改善. | [11] |
| Bmal1ノックアウトマウス | 500 mg/kg | 腹腔内 | 10日間 | 肝臓全体およびミトコンドリアNAD+量を増加させ,SIRT3標的タンパクの脱アセチル化を亢進し,ミトコンドリア呼吸能および脂肪酸酸化を改善. | [10] |
| 野生型若年マウス(6月齢) | 300 mg/kg | 経口 | 12か月間 | 老化に伴う海馬の神経幹細胞の減少が抑制され,新生神経が増加傾向. | [16] |
| 野生型老年マウス(30月齢) | 500 mg/kg | 腹腔内 | 7日間 | SIRT2の活性化を介して,寿命延長効果をもたらす可能性. | [20] |
| Nampt+/−雌マウス,老化した膵β細胞特異的SIRT1トランスジェニックマウス | 500 mg/kg | 腹腔内 | 腹腔内糖負荷試験 14時間前 | 糖刺激に対するインスリン分泌促進表現型の回復. | [1][2][4] |
| NR | 高脂肪食負荷誘導性肥満マウス | 400 mg/kg | 経口 | 7日間 | 肝臓,褐色脂肪組織,骨格筋のNAD+量が増加し,SIRT1およびSIRT3が触媒する反応を促進させることで,高脂肪食に合併する体重増加,インスリン抵抗性,脂質異常症などの代謝異常症を改善. | [8] |
| Sco2 knockout/knockinマウス(ミトコンドリア病モデルマウス) | 400 mg/kg | 経口 | 4週間 | ミトコンドリア呼吸鎖が改善し,運動耐用能が上昇. | [12] |
| 肝細胞特異的URI過剰発現マウス | 500 mg/kg | 経口 | 最大48週間 | 肝臓のNAD+量を増加させ,DNA傷害・腫瘍形成を抑制. | [18] |
| アルツハイマー病モデルマウス | 250 mg/kg | 経口 | 12週間 | 大脳皮質NAD+量を増加させ,βアミロイド産生が低下することで認知機能が改善. | [14] |
| Wallerian degeneration slow (wlds) mice | 1000 mg/kg | 腹腔内 | 1日2回5~19日 | 蝸牛のNAD+量を増加させ,SIRT3が触媒する反応の促進を介して,騒音性難聴およびらせん神経節細胞変性を抑制. | [15] |
| ミトコンドリア筋症マウス | 400 mg/kg | 経口 | 16週間 | 骨格筋・褐色脂肪組織におけるミトコンドリア合成の促進.ミトコンドリア形態異常,ミトコンドリアDNA欠失を抑制し病態進展を抑制. | [13] |
1)NAD+合成系
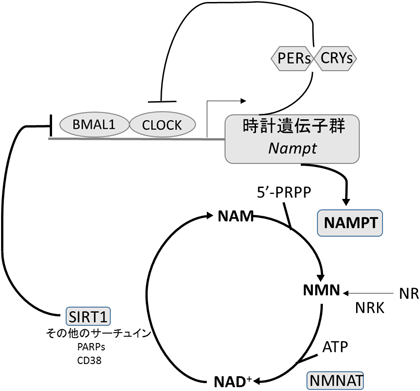
NAD+合成の材料はトリプトファン,および水溶性ビタミンB3として総称されるニコチンアミド(nicotinamide: NAM)とニコチン酸(nicotinic acid: NA),そしてNRが知られている.哺乳類はNAMを主要材料とし,そこから2段階の酵素反応を経てNAD+を合成する.第一段階では,NAMPTがNAMを5′-ホスホリボシル-1′-ピロリン酸(5′-phophoribosyl-1′-pyrophosphate: PRPP)の存在下にNMNに変換する.続いてNMNはATPと,第二の酵素であるニコチンアミド/ニコチン酸モノヌクレオチドアデニリルトランスフェラーゼ(nicotinamide/nicotinic acid mononucleotide adenylyltransferase: NMNAT)によって,NAD+へと合成される.また,NRはニコチンアミドリボシドキナーゼ(nicotinamide riboside kinase: NRK)によりNMNへと変換され,NAD+が合成される1–3)(図1).第一の酵素であるNAMPTは,二量体を形成するⅡ型ホスホリボシルトランスフェラーゼに属し,NAMへの基質特異性がきわめて高く(Km=0.92 µM),哺乳類NAD+合成系における律速酵素として機能する.NAMPTには細胞内型NAMPT(intracellular NAMPT: iNAMPT)と細胞外型NAMPT(extracellular NAMPT: eNAMPT)の二つの型が存在する.iNAMPTについては後述するが,eNAMPTは,成熟した脂肪細胞,肝細胞,白血球細胞,心筋細胞から分泌され,サイトカイン,ホルモン様に血中を巡ることが報告されている.特に成熟脂肪細胞から分泌されるeNAMPTはiNAMPTより高い酵素活性を有し,細胞外コンパートメントにおけるNMNの合成を介して,全身性にNAD+合成系を調節することが示唆されており2,4),eNAMPTの生理学的意義に関して今後の詳細な解析結果が待たれる.
2)NAD+消費酵素
哺乳類において合成されたNAD+は,ポリADPリボースポリメラーゼ(poly ADP ribose polymerases: PARPs),NAD+グリコヒドロラーゼ/ADPリボシルシクラーゼ(CD38ファミリー),サーチュインなどの主なNAD+消費酵素によって,ADPリボシル化反応,脱アセチル化反応などの酵素反応基質として使用される1–3).PARPsやCD38の阻害によりNAD+濃度が増加することで,サーチュインの触媒する反応が促進される1).
i)PARPs, CD38
PARPsは,DNA損傷に伴い活性化され,NAD+を基質として核タンパク質にADPリボシル基を転移・重合する翻訳後修飾を行い,主にPARP-1およびPARP-2がDNA修復,細胞死誘導,エネルギー代謝など,分子機能や細胞機能において重要な役割を果たしている.PARP-1ノックアウトマウスでは,筋肉と褐色脂肪組織においてNAD+量が増加し,SIRT1の触媒する反応の促進が認められ,野生型マウスに比して,ミトコンドリア機能,エネルギー消費が促進され,耐糖能も改善し,体脂肪量も低下する1).
CD38は多機能酵素で膜貫通型タンパク質であり,NAD+から細胞内Ca2+動員メッセンジャーであるサイクリックADPリボース合成とADPリボースへの加水分解を触媒する.そのため,CD38ノックアウトマウスでは主要臓器で著明な組織NAD+量の増加を認め,サーチュインの触媒する反応が促進され,高脂肪食負荷に合併する肥満やメタボリック症候群に対して保護的に働く1,3).これらの結果は,PARPs,CD38阻害が,エネルギー代謝やミトコンドリア機能を改善する創薬標的となりうることを示唆している.実際に,PARP-1およびPARP-2の阻害薬であるparibs,CD38阻害薬であるapigeninは,代謝機能を改善させることが報告されている1,3).
ii)サーチュイン
哺乳類では七つのサーチュイン(SIRT1~7)が存在し5),中でもSIRT1の解析が最も進んでいる.近年,脳特異的SIRT1過剰発現マウス(BRASTO)で,視床下部背内側核,外側核において神経活動性が亢進し,寿命延長効果が得られることが報告されたことは記憶に新しい1).SIRT1によって触媒される段階の速度はNAMPTによるNAD+合成を介して直接的に制御されている1–3).さらにNAMPTを介したNAD+合成系とSIRT1は生体の概日リズム形成に重要な役割を果たすことが明らかにされた1,2,6).具体的には,NAMPTおよびNAD+が概日リズムを刻み,NAD+濃度の変化によりSIRT1の触媒する反応速度が変化する.SIRT1は時計遺伝子の転写ネットワークを制御するCLOCK:BMAL1とクロマチン複合体を形成し,BMAL1を脱アセチル化することで,CLOCK:BMAL1の転写活性を抑え概日リズムをコントロールする(図1).これらの知見は,代謝,サーカディアンリズム,老化,という独立した生命現象のクロストークにおけるNAMPT-NAD+-SIRT1の重要性を示唆するものといえる.
3. 哺乳類NAD+合成系の老化関連疾患における役割と治療標的としての可能性
近年,NAMPTを中心としたNAD+合成系,NAD+消費酵素に関する研究は爆発的な勢いで展開され,その結果,特に老化関連疾患における病態生理学的重要性が次々と解明されている.そして同時に,種々のモデル動物において,NMN,NRに代表されるNAD+中間代謝産物の多彩な効能が報告されている(表1).ここでは,主として老化関連疾患における,哺乳類NAD+合成系の役割と治療標的としての可能性を最新の知見をもとに考察したいと思う.
1)代謝疾患
老化・加齢は,インスリン抵抗性,膵β細胞機能異常の最も重要なリスクファクターとして知られている.膵臓,白色脂肪組織,骨格筋などの主要代謝臓器においては,老年マウス(25~31月齢)では若年マウス(3~6月齢)に比べて,NAMPTタンパク質,NAD+量が低下していることが発見された.同時に高脂肪食負荷誘導性糖尿病マウスでは通常食マウスに比して,肝臓,白色脂肪組織中のiNAMPTタンパク質量,NAD+量がともに低下することも明らかにされた7).重要なことに,NAMPTの酵素反応産物であるNMN(500 mg/kg)の腹腔内投与は,NAD+合成系を回復し,SIRT1の触媒する反応が促進され,高脂肪食負荷,老化に合併する耐糖能異常,インスリン抵抗性,脂質異常症を改善した7).さらにNRも経口投与すると肝臓,褐色脂肪組織,骨格筋のNAD+量を増加させ,SIRT1およびSIRT3の触媒する反応を促進させることで,高脂肪食負荷に合併する体重増加,インスリン抵抗性,脂質異常症などの代謝異常症を改善することが報告されている8).近年,これらNAD+中間代謝産物のミトコンドリア機能への効用が注目されている.たとえば,NMNは,加齢に伴う骨格筋のミトコンドリア機能不全を劇的に回復させ,骨格筋萎縮や炎症のマーカー,インスリンシグナル伝達,グルコースの取り込みを改善する9).また,時計遺伝子Bmal1ノックアウトマウスにおいて,NMNは肝臓全体およびミトコンドリアNAD+量を増加させ,ミトコンドリアサーチュインSIRT3の標的タンパク質の脱アセチル化を亢進し,ミトコンドリア呼吸能および脂肪酸酸化を改善する10).そして,心筋でミトコンドリア呼吸鎖複合体Iの機能を低下させたマウスにおいても,NMNは,心筋におけるNAD+/NADH比を上昇させ,ミトコンドリアタンパク質の脱アセチル化を亢進し,ミトコンドリア膜透過性遷移孔の透過性を改善させる11).さらに近年,シトクロムcオキシダーゼ合成が障害されたミトコンドリア病モデルであるSco2 knockout/knockinマウスでは,NRの経口投与により,NAD+量の増加およびFOXO1の脱アセチル化の亢進を認め,ミトコンドリア呼吸鎖が改善し,運動耐用能が上昇することが報告されている12).それに合致して,NRの投与により,ミトコンドリア筋症を呈するマウスにおいて,骨格筋および褐色脂肪組織におけるミトコンドリア合成が促進され,ミトコンドリア形態異常やミトコンドリアDNA欠失を抑制し病態進展が抑制された13).老化の病態生理におけるミトコンドリアの重要性を考慮すると9),今後,臓器特異的に,NAMPT-NAD+合成系がミトコンドリア機能に果たす役割を詳細に解析し,創薬標的としての可能性をさらに探索していくことが,この研究領域の最重要課題の一つといえる.
2)神経疾患
近年,神経領域におけるNAD+合成系やサーチュインに関する研究は特に注目されている1).アルツハイマー病モデルマウスにNR(250 mg/kg/day)を3か月間経口投与したところ,大脳皮質NAD+量が増加し,peroxisome proliferator-activated receptor-γ coactivator 1α(PGC-1α)の発現亢進に伴いアミロイド前駆体タンパク質(APP)切断酵素βセクレターゼ(BACE1)が分解され,βアミロイド産生が低下することで認知機能が改善した14).また,NR(1000 mg/kg)を投与することにより蝸牛のNAD+が上昇し,SIRT3の触媒する反応を促進させ,騒音性難聴およびらせん神経節細胞変性が抑制された15).さらに,6月齢から18月齢までNMN(300 mg/kg)を経口投与したマウスでは,老化に伴う海馬の神経幹細胞の減少が抑制され,新生神経の増加傾向を認めた16).近年,神経変性疾患や神経障害のげっ歯類モデル動物において,aminopropyl carbazoleの一つであり神経細胞死を抑制するP7C3はNAMPTと結合することで酵素活性を上げ,NAD+量の増加を介して細胞保護・外傷性脳損傷後の軸索変性を予防することが解明され17),創薬対象としてのNAMPTの重要性が認識されている.
3)がん
最近,NAD+合成系と発がんの関連を検討した興味深い研究成果が発表された.具体的には,L-トリプトファン/キヌレニンを代謝してNAD+を産生する系の酵素の転写を抑制するunconventional prefoldin RPB5 interactor(URI)を肝細胞特異的に過剰発現したマウスでは,DNA傷害により段階的に肝細胞がんへと進展した.NAD+中間代謝産物であるNR(500 mg/kg/day)を経口投与することにより,肝臓におけるNAD+量が増加し,DNA傷害・腫瘍形成が抑制されたことから,NAD+量増加による発がん抑制の可能性が示された18).しかしその一方で,NAMPTの特異的酵素阻害剤(FK866)によりNAD+濃度を低下させると,グリセルアルデヒド-3-リン酸デヒドロゲナーゼの酵素反応が低下することによりがん細胞が依存する解糖系が抑制され,がんの進展抑制作用があることが知られている.今後,がんの病態形成におけるNAD+合成系の役割に関するより詳細な検討が待たれる.
4)腎臓疾患
腎機能は加齢に伴い低下するため,慢性腎臓病は一種の老化疾患と考えられる.近年,腎臓疾患におけるNAD+合成系およびサーチュインの役割も次々と解明されている.たとえば,最近,糖尿病性腎症発症における近位尿細管と糸球体の連関にNMNが重要な役割を果たしていることが報告された.糖尿病では,最初に近位尿細管Sirt1が低下することにより,近位尿細管から糸球体に放出されるNMNが低下する.さらに,NMNの減少で足細胞Sirt1も低下し,糸球体のタイトジャンクションであるClaudin-1の発現をエピジェネティックに調節し,糸球体基底膜のスリット膜タンパク質の異常を引き起こし,蛋白尿が出現することが報告された.これらの病理所見は,近位尿細管特異的Sirt1過剰マウスで軽減し,それとは逆に欠損マウスで増悪した19).このようにNAD+中間代謝産物,SIRT1が糸球体・尿細管に対し保護的に働くことが示唆され,老化による慢性腎臓病の進展抑制に向けての治療標的として期待される.
近年の研究成果により,老化関連疾患におけるNAD+合成系の重要性が明らかにされ,NMNやNRなどのNAD+中間代謝産物が老化関連疾患の創薬標的として期待されている(図2,表1).興味深いことに最近,脳特異的SIRT1過剰発現マウス(BRASTO)および全身性SIRT6過剰発現マウスに寿命延長効果が得られることが報告された1).また,老化に伴うNAD+の低下がSIRT2の活性低下を引き起こし寿命が短くなる一方で,NMN投与がSIRT2の活性化を介して寿命延長効果をもたらす可能性が示唆された20).これらの結果は,NAD+中間代謝産物投与により,サーチュインを活性化することで,寿命延長効果を発揮する可能性をも示唆しており,今後はヒトへの臨床応用の早期実現を目指して研究が展開されていくと考えられる.我々のグループでは現在,遺伝子操作動物を駆使して,脂肪細胞のミトコンドリア機能におけるNAD+合成系の生理学的重要性を解析するとともに,ヒトの代謝機能(特にインスリン感受性),サーカディアンリズム制御におけるNAMPT-NAD+-SIRT1の役割を検討する臨床研究を鋭意展開している.今後,基礎・臨床研究が一体となったトランスレーショナル型リサーチを発展させていくことで,超高齢社会において今後も増加が予想される老化関連疾患に対する新規治療法の開発が期待される.
謝辞Acknowledgments
長年ご指導いただいております慶應義塾大学医学部内科の伊藤裕教授,ワシントン大学医学部発生生物学部門の今井眞一郎教授にこの場を借りて厚く御礼申し上げます.また,著者(山口慎太郎)の留学に際してご支援をいただいた住友生命福祉文化財団に厚く御礼申し上げます.紙面の都合上一部の論文しか引用できなかったことをお詫び致します.
引用文献References
1) Imai, S. & Guarente, L. (2014) Trends Cell Biol., 24, 464–471.
2) Imai, S. & Yoshino, J. (2013) Diabetes Obes. Metab., 15(Suppl 3), 26–33.
3) Yoshino, J. & Imai, S. (2013) Methods Mol. Biol., 1077, 203–215.
4) Revollo, J.R., Korner, A., Mills, K.F., Satoh, A., Wang, T., Garten, A., Dasgupta, B., Sasaki, Y., Wolberger, C., Townsend, R.R., Milbrandt, J., Kiess, W., & Imai, S. (2007) Cell Metab., 6, 363–375.
5) Nakagawa, T. & Guarente, L. (2014) Cell Metab., 20, 192–192.
6) Ramsey, K.M., Yoshino, J., Brace, C.S., Abrassart, D., Kobayashi, Y., Marcheva, B., Hong, H.K., Chong, J.L., Buhr, E.D., Lee, C., Takahashi, J.S., Imai, S., & Bass, J. (2009) Science, 324, 651–654.
7) Yoshino, J., Mills, K.F., Yoon, M.J., & Imai, S. (2011) Cell Metab., 14, 528–536.
8) Canto, C., Houtkooper, R.H., Pirinen, E., Youn, D.Y., Oosterveer, M.H., Cen, Y., Fernandez-Marcos, P.J., Yamamoto, H., Andreux, P.A., Cettour-Rose, P., Gademann, K., Rinsch, C., Schoonjans, K., Sauve, A.A., & Auwerx, J. (2012) Cell Metab., 15, 838–847.
9) Gomes, A.P., Price, N.L., Ling, A.J., Moslehi, J.J., Montgomery, M.K., Rajman, L., White, J.P., Teodoro, J.S., Wrann, C.D., Hubbard, B.P., Mercken, E.M., Palmeira, C.M., de Cabo, R., Rolo, A.P., Turner, N., Bell, E.L., & Sinclair, D.A. (2013) Cell, 155, 1624–1638.
10) Peek, C.B., Affinati, A.H., Ramsey, K.M., Kuo, H.Y., Yu, W., Sena, L.A., Ilkayeva, O., Marcheva, B., Kobayashi, Y., Omura, C., Levine, D.C., Bacsik, D.J., Gius, D., Newgard, C.B., Goetzman, E., Chandel, N.S., Denu, J.M., Mrksich, M., & Bass, J. (2013) Science, 342, 1243417.
11) Karamanlidis, G., Lee, C.F., Garcia-Menendez, L., Kolwicz, S.C. Jr., Suthammarak, W., Gong, G., Sedensky, M.M., Morgan, P.G., Wang, W., & Tian, R. (2013) Cell Metab., 18, 239–250.
12) Cerutti, R., Pirinen, E., Lamperti, C., Marchet, S., Sauve, A.A., Li, W., Leoni, V., Schon, E.A., Dantzer, F., Auwerx, J., Viscomi, C., & Zeviani, M. (2014) Cell Metab., 19, 1042–1049.
13) Khan, N.A., Auranen, M., Paetau, I., Pirinen, E., Euro, L., Forsstrom, S., Pasila, L., Velagapudi, V., Carroll, C.J., Auwerx, J., & Suomalainen, A. (2014) EMBO Mol. Med., 6, 721–731.
14) Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., Ho, L., Wang, J., Sastre, M., Ono, K., Sauve, A.A., & Pasinetti, G.M. (2013) Neurobiol. Aging, 34, 1581–1588.
15) Brown, K.D., Maqsood, S., Huang, J.Y., Pan, Y., Harkcom, W., Li, W., Sauve, A., Verdin, E., & Jaffrey, S.R. (2014) Cell Metab., 20, 1059–1068.
16) Stein, L.R. & Imai, S. (2014) EMBO J., 33, 1321–1340.
17) Wang, G., Han, T., Nijhawan, D., Theodoropoulos, P., Naidoo, J., Yadavalli, S., Mirzaei, H., Pieper, A.A., Ready, J.M., & McKnight, S.L. (2014) Cell, 158, 1324–1334.
18) Tummala, K.S., Gomes, A.L., Yilmaz, M., Grana, O., Bakiri, L., Ruppen, I., Ximenez-Embun, P., Sheshappanavar, V., Rodriguez-Justo, M., Pisano, D.G., Wagner, E.F., & Djouder, N. (2014) Cancer Cell, 26, 826–839.
19) Hasegawa, K., Wakino, S., Simic, P., Sakamaki, Y., Minakuchi, H., Fujimura, K., Hosoya, K., Komatsu, M., Kaneko, Y., Kanda, T., Kubota, E., Tokuyama, H., Hayashi, K., Guarente, L., & Itoh, H. (2013) Nat. Med., 19, 1496–1504.
20) North, B.J., Rosenberg, M.A., Jeganathan, K.B., Hafner, A.V., Michan, S., Dai, J., Baker, D.J., Cen, Y., Wu, L.E., Sauve, A.A., van Deursen, J.M., Rosenzweig, A., & Sinclair, D.A. (2014) EMBO J., 33, 1438–1453.
21) Yamamoto, T., Byun, J., Zhai, P., Ikeda, Y., Oka, S., & Sadoshima, J. (2014) PLoS ONE, 9, e98972.
著者紹介Author Profile
 山口 慎太郎(やまぐち しんたろう)
山口 慎太郎(やまぐち しんたろう)ワシントン大学医学部内科博士研究員(吉野純研究室).医学博士(M.D., Ph.D.).
略歴2005年慶應義塾大学医学部卒業.13年同大学院医学研究科博士課程修了,医学博士(内科学).13~14年同大学医学部内科学教室助教(腎臓内分泌代謝内科,伊藤裕教授).14年より現職.
研究テーマと抱負ヒトの代謝機能,サーカディアンリズム制御におけるNAMPT-NAD+-SIRT1の役割を検討すべくトランスレーショナル型リサーチを展開している.
 吉野 純(よしの じゅん)
吉野 純(よしの じゅん)ワシントン大学医学部内科Assistant Professor. 医学博士(M.D., Ph.D.).
略歴2000年慶應義塾大学医学部卒業,04年同大学院医学研究科博士課程修了,医学博士(内科学).04~07年同大学医学部内科学教室助手(腎臓内分泌代謝内科,伊藤裕教授),内科学研修医・専修医課程修了.07~12年米国ワシントン大学医学部発生生物学部部門博士研究員(今井眞一郎研究室).12年より現職.
研究テーマと抱負ヒトにおけるインスリン抵抗性の分子メカニズムの解明を目指したトランスレーショナル型リサーチ.特に,脂肪細胞におけるNAD合成系,時計遺伝子に注目して研究を展開中.