酵素反応の多くは補酵素を必要とする.多くの補酵素はそれ自体が触媒機能を持ち,単独でも反応を触媒できるが,酵素タンパク質の活性中心に結合した場合と比べると,その反応速度ははるかに低く,かつ反応特異性が低下し副反応を触媒してしまう.補酵素としてのピリドキサール5′-リン酸(pyridoxal 5′-phosphate: PLP)はその典型的な例といえる.PLPはアミノ酸のアミノ基転移反応,脱炭酸反応,脱離反応,アルドール開裂,クライゼン型縮合反応などを触媒する多機能触媒である.PLPを補酵素とする酵素(以下,PLP酵素)は,それぞれに特有のタンパク質構造が機能することによって,触媒の各素過程において特定の反応だけが進行するよう精密に化学反応を制御している.それゆえに,野生型酵素における通常の反応解析において,抑制されている副反応が検出されることはほとんどない.本稿では,スフィンゴ脂質生合成経路のkey enzymeであるセリンパルミトイル転移酵素(serine palmitoyltransferase: SPT)の活性発現機構の研究過程で,変異酵素で進行した副反応の解析を通して明らかとなった本酵素の反応制御機構について述べる.
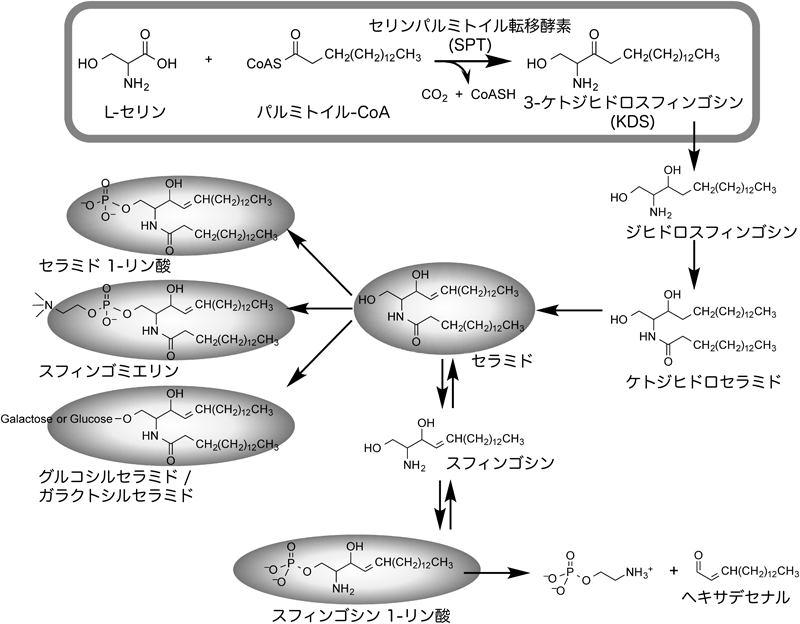
セラミドに代表されるスフィンゴ脂質は細胞間,細胞内の情報伝達物質として,また,形質膜における脂質ラフト(lipid raft)の構成成分として,多彩な生理機能を有する脂質である1).スフィンゴ脂質生合成はL-セリンとパルミトイル-CoAの縮合・脱炭酸反応による長鎖塩基3-ケトジヒドロスフィンゴシン(3-ketodihydrosphingosine: KDS)の生成で開始される(図1).この反応はスフィンゴ脂質生合成の律速段階であり,PLP酵素であるSPTによって触媒される2).KDSの3位ケトン基がヒドロキシ基に還元された後,アシル基転移,Δ4不飽和化によってセラミドに変換される.セラミドのヒドロキシメチル基に対するリン酸化,ホスホコリン付加,糖鎖付加によってセラミド1-リン酸,スフィンゴミエリン,および種々のスフィンゴ糖脂質に変換される.セラミドがセラミダーゼによってスフィンゴシンに変換され,これがリン酸化されるとスフィンゴシン1-リン酸を生じる.スフィンゴ脂質代謝物は細胞の増殖・分化・運動性などに関わる脂質メディエーターであり,また,形質膜上で脂質ラフトと呼ばれるマイクロドメインを形成し,種々の情報伝達や膜輸送を介する特定の物質輸送の場としても機能する.スフィンゴ脂質ホメオスタシスの破綻は細胞に深刻なダメージを与えることから,スフィンゴ脂質生合成に関わる酵素には副反応抑止のための厳密な制御機構が存在する.
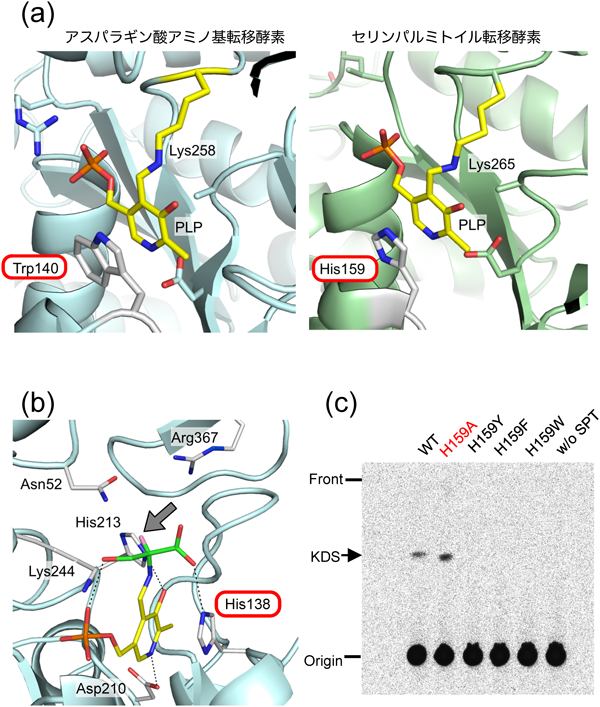
真核生物SPTは取り扱いの難しい膜結合型タンパク質であり,立体構造解析や詳細な反応解析は現在でも困難である3).一方,細菌由来SPTは水溶性で4–6),真核生物SPTの原型として分子レベルでの詳細な研究を可能にする強力なモデル系である.著者らは細菌由来SPTを実験対象として酵素反応の速度論的解析と立体構造解析に取り組んできた4–12).構造学的特徴から,SPTはフォールドタイプIのPLP酵素に分類される.このグループのPLP酵素の多くにおいては,PLP–リシン分子内Schiff塩基のre面側に,PLPピリジン環に重なるように芳香族アミノ酸残基が配置される(図2a左)13).一方,SPTはこの位置にヒスチジン残基を有し(図2a右),このヒスチジン残基がSPTの反応機構におけるユニークな役割を果たすと予想された10).
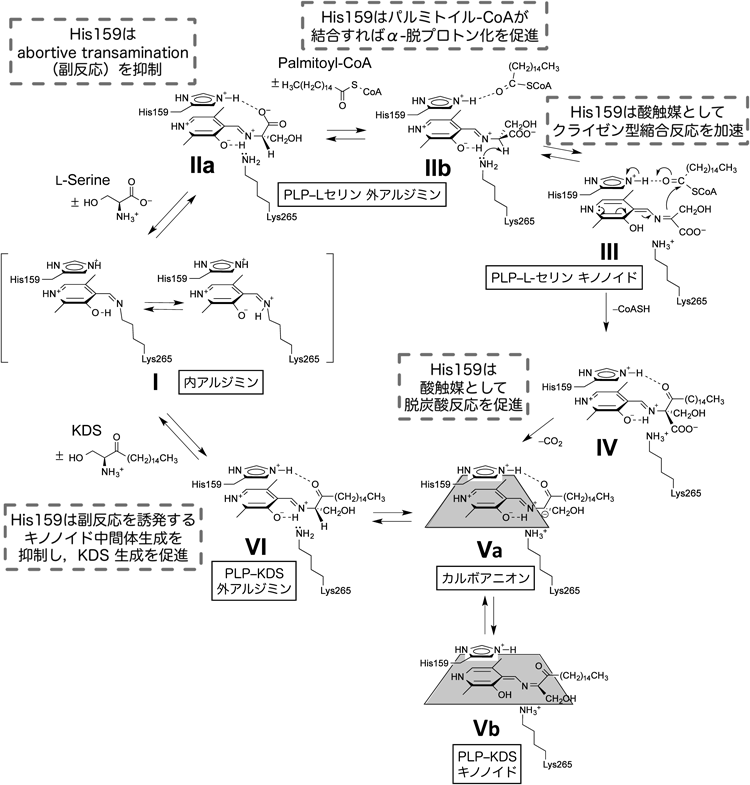
SPTの反応機構の概略を図3に示す.SPTの活性中心において,PLPはLys265のε-アミノ基との間にアルジミンを形成する(内アルジミンinternal aldimine, I).一つめの基質であるL-セリンの結合によってイミノ基交換反応が進行してPLP–L-セリンアルジミン(外アルジミンexternal aldimine, II)が生じる.二つめの基質であるパルミトイル-CoAの結合後にα-脱プロトン化が進行して一つめのキノノイド中間体(III)となり,これのカルボアニオン性Cαがパルミトイル-CoAを攻撃して縮合生成物(IV)が生じる(クライゼン型縮合反応).縮合生成物の脱炭酸(V)とCα位へのプロトン付加によってKDSが生成し(PLP–KDSアルジミン中間体,VI),酵素からKDSが解離して内アルジミンIが再生され,反応が完結する.著者らは,Sphingomonas paucimobilis由来SPTの反応をさまざまな基質アナログを用いて解析し,L-セリンからのα-プロトンの引き抜きによるキノノイド中間体(III)の生成過程がSPT触媒反応の律速段階であることを速度論的に証明した8).並行してSphingobacterium multivorum由来SPTについてL-セリンとの複合体の結晶化に成功し立体構造を決定したところ(図2b),SPT活性中心ではL-セリンがアミノ基でPLPと外アルジミン中間体(図3,IIa)を形成し,L-セリンのカルボキシ基は2個の水分子に加えてHis138の側鎖Nε2と水素結合していることが明らかになった9,10).Sphingomonas SPTについてもL-セリン複合体の構造解析から活性部位における同様の水素結合が確認された14,15).SPTにおいて,活性部位でPLPに重なるように配置されたヒスチジン残基はL-セリンとパルミトイル-CoAの両基質のアンカー部位であり,触媒反応後半の素過程にも直接関与する可能性が示唆された8).しかしながら,野生型酵素を用いた解析からは,この仮説を裏づけるような,また,ヒスチジン残基の役割についてそれ以上の考察を可能にするような実験データは得られなかった.
4. His159はSPT活性の発現に必須ではない
触媒基としての役割を確かめるためにSphingomonas SPTのHis159をアラニン,フェニルアラニン,チロシン,トリプトファン残基に置換した4種類の変異型酵素(H159A, H159F, H159Y, H159W)を調製し,酵素反応を解析した11).[14C]-L-セリンを基質として反応させたところ,H159Aにおいてのみ[14C]-KDSが検出された(図2c).これは,His159の側鎖がSPTの酵素活性「それ自体」には必須ではないことを意味しており,予想に反する結果であった.一方,芳香族アミノ酸残基へ置換した変異型酵素については反応生成物が検出されず,芳香族アミノ酸残基ではHis159の機能が代替されないことが示された.定常状態下の速度論的解析の結果,H159Aは野生型酵素の16%(野生型酵素kcat=0.69 s−1,H159A kcat=0.11 s−1)の活性を維持していることが判明した.H159Aでは野生型酵素と比較してL-セリンに対するKm値は約10倍上昇していたが(野生型酵素Km=6.2 mM, H159A Km=58.1 mM),パルミトイル-CoAに対するKm値には変異の影響がほとんど現れなかった(野生型酵素Km=1.0 mM, H159A Km=0.72 mM).以上の結果は,His159がL-セリンの結合部位として機能することを明確に示すものである.
5. His159はSPTの反応特異性を決定づけている
SPTは補酵素PLPに由来する特徴的な吸収スペクトルを示し,その変化は活性中心における基質分子の化学的な状態変化を鋭敏に反映する.精製標品と基質の反応や,中間体を模した状態で反応停止するような基質アナログとの反応をさまざまな分光学的手法で解析することにより,SPT触媒反応の素過程を詳細に調べることが可能である.
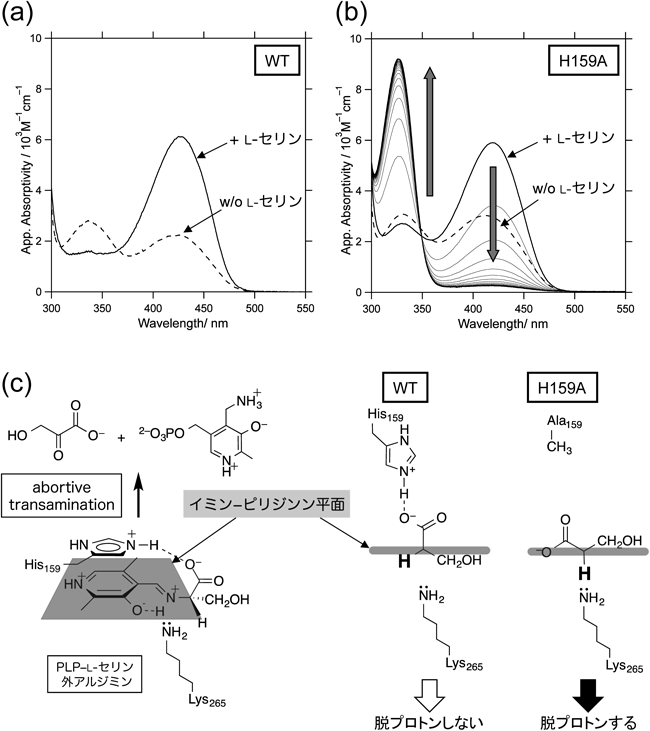
野生型SPTに基質のL-セリンを添加すると,ミカエリス複合体形成とそれに続くSchiff塩基の交換反応によってPLP–L-セリン外アルジミン中間体(図3のIIa)を生成する.基質が結合する前のSPTは,活性部位のPLPがリシン残基のε-アミノ基との間で分子内Schiff塩基(内アルジミン;図3のI)を形成しており,420 nm付近と340 nm付近に二つの極大を持つ図4aの破線のような吸収スペクトルを示すが,加えたL-セリンの濃度に依存して420 nm付近の吸収ピークが増加し,340 nm付近のピークが減少する(図4a,実線).
H159AにL-セリンを添加すると,いったんは野生型酵素と同様に420 nm付近の吸収の増加と340 nm付近の吸収の減少が観察され,PLP–L-セリンアルジミン中間体の生成が確認された(図4b).ところが,このスペクトルは不安定で時間経過に伴って420 nmの吸収は徐々に減少し,330 nmに新しいピークが現れた.スペクトル変化完了後に酵素タンパク質を限外濾過によって除いた試料について高速液体クロマトグラフィーで分析したところ,PLPが当量のピリドキサミンリン酸(pyridoxamine 5′-phosphate: PMP)へ変化していることが判明した.このことは,本来ならSPTが触媒しないアミノ基転移反応が進行して酵素が不活化したことを意味する(図4c).PLP酵素におけるこのようなアミノ基転移反応は好ましくない副反応であり,目的の生成物を作らないことから,「実を結ばないアミノ基転移反応」ということで“abortive transamination”と呼ばれる.この反応の速度定数は4.43×10−4 s−1と見積もられた(表1).他の変異型SPTについてもL-セリン添加によってH159Aと同様の現象が観察された.野生型SPT–L-セリン複合体の結晶構造においてL-セリン部分のカルボキシ基がHis159によって固定されていることと10,15),野生型酵素とL-セリンの反応においてabortive transaminationが観察されないことを考え合わせると,変異型酵素における観測結果は下記のように解釈された.野生型酵素においては,図4cで示すように,His159–L-セリン間の水素結合によってアルジミンCαとカルボキシ基の炭素原子間の結合はイミン–ピリジン平面に対して垂直になる.その結果,アルジミンのL-セリン部分のCα–H結合はイミン–ピリジン平面から30°ずれ,脱プロトン化に適さないコンホメーションとなる.しかし変異型酵素においては,相互作用するアミノ酸側鎖が存在しないためにCα–N結合を軸とする自由な回転が可能になり,Cα–H結合がイミン−ピリジン平面に対して垂直になったときにα-脱プロトン反応が進行すると考えられる.結合の回転の自由度の増加と裏腹に,常に最適な配向に固定されるわけではないため,変異型SPTのabortive transaminationの速度定数[(0.6~4.4)×10−4 s−1]は天然のアミノ基転移酵素の速度定数(数百~数千s−1)よりはるかに低い.しかし,変異型SPTで観察されたこの副反応は,天然のアミノ基転移酵素のいわゆる半反応に該当することから,His159を変異させることによってSPTの反応特異性が変化したとも解釈できる現象である.
表1 PLPおよびL-セリンとの相互作用に関するSPTの解離定数と副反応の速度定数 | PLP | L-セリン | abortive transamination |
|---|
| Kd (µM) | Kd (mM) | k (s−1) |
|---|
| WT | 27.3±2.6 | 1.4±0.1 | ND |
| H159A | 18.9±3.0 | 77.1±14.4 | (4.43±0.02)×10−4 |
| H159F | 3.0±0.4 | 27.8±2.0 | (11.67±0.03)×10−5 |
| H159Y | 12.9±3.6 | 19.8±1.5 | (6.34±0.04)×10−5 |
| H159W | 17.3±3.1 | 21.6±1.5 | (6.75±0.03)×10−5 |
PLPとL-セリンに対する解離定数,abortive transaminationの反応速度定数を表1にまとめた.すべての変異体において,PLPに対するKdは野生型酵素の値と大差がみられないことから,His159はPLPの結合に対しては大きく寄与しないと考えられる.一方でL-セリンに対するKdは,野生型SPTのKd=1.4 mMに対して,H159AではKd=77.1 mMと55倍の増加を示した.His159を芳香族アミノ酸残基へ置換した変異型SPTではそれぞれKd=27.8 mM,19.8 mM,21.6 mMであり,野生型酵素より10~20倍増加していた.以上の結果から,His159はL-セリンの結合部位として重要であり,SPTの反応特異性を決定づけている残基であることが明らかになった.
6. H159Aについての遷移状態の反応速度論的解析―パルミトイル–CoAの結合によるα-脱プロトン化
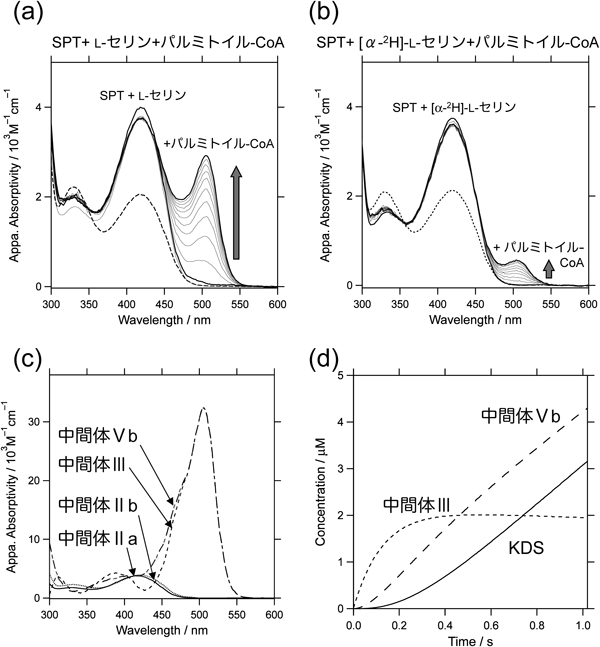
SPT変異体のうち,H159Aのみが酵素活性を維持していたことから,H159A SPT–L-セリン複合体においてabortive transaminationが進行する前にパルミトイル-CoAと混合できるように構成したストップトフロー装置を用いて,SPTとL-セリン,パルミトイル-CoAの反応について時間分解スペクトルを測定し,遷移状態の速度論的解析を行った.H159Aではパルミトイル-CoAとの混合によって505 nmに顕著な吸収増加が観測された(図5a).このようなスペクトルは,野生型酵素やHis159を芳香族アミノ酸残基に置換した変異型酵素では観測されなかった.
さらに,スペクトル分解による解析の結果,二つの中間体の存在が示唆され,これを満たす様式として下記のモデルを想定した:
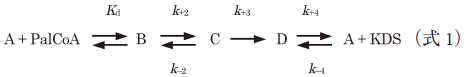
ここで,Aは酵素–L-セリン二者複合体,Bは酵素–L-セリン–パルミトイル-CoA三者複合体を示す.CとDは中間体である.SPT–L-セリン二者複合体にパルミトイル-CoAが結合して三者複合体になり,二つの中間体を経由して生じた生成物KDSが解離する機構を想定している.実際のストップトフロー実験系では酵素–L-セリン二者複合体溶液とパルミトイル-CoA溶液を迅速混合するので,基質フリーのSPTには飽和量存在するL-セリンが速やかに結合して酵素–L-セリン二者複合体が再生するモデルになる(式1の右辺のA).このモデルに基づいて時間分解スペクトルのグローバル解析を行い,各過程の反応速度定数と反応中間体の理論スペクトルを算出した.グローバル解析とは多波長同時に時間分解測定したデータを多変量解析し,想定した反応中間体の生成消滅の過程を追跡する解析手法である.求められた反応中間体C, Dの理論スペクトルはいずれも500 nm付近に30,000 M−1 cm−1以上のモル吸光係数を持つキノノイドの化学構造に特徴的なピークを有し,他のPLP酵素で観測される一般的なキノノイド中間体の吸収スペクトルの形状とよく一致した(図5c).よってCとDはキノノイド中間体であり,図3における中間体IIIと中間体Vbに対応すると考えられた.
同位体効果が顕著に現れる過程を検出することを目的として,重水素化した[α-2H]-L-セリンを用いて同様の速度論的解析を行った.通常のL-セリンを用いた場合と比較すると,観察されたキノノイド中間体の蓄積は少なかったが,時間分解スペクトルのグローバル解析によって得られた中間体のスペクトルは本質的に同様であった.算出された反応速度定数の中で,k+2が[α-2H]-L-セリンに対して0.064 s−1,L-セリンに対して0.46 s−1と高い速度論的同位体効果を示した(表2).同位体効果の値7.2はアミノ基転移酵素のCα位脱プロトン化についての文献値ともよく一致した16).この結果は,k+2がL-セリンのCα位の脱プロトン化の速度定数であることを示し,キノノイド中間体(式1のC)がPLP–L-セリンアルジミン中間体の脱プロトン化によって生成することを強く支持するものである(図3, III).グローバル解析によって得られた反応動力学定数を表2と下記の式2,式3にまとめた.
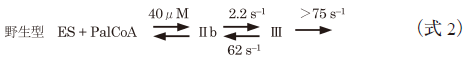
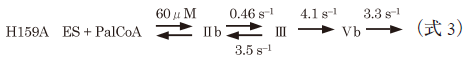
ここで,ESはSPT–L-セリン二者複合体,PalCoAはパルミトイル-CoAであり,IIb,IIIおよびVbは図3で示す反応中間体に該当する.H159Aのα-脱プロトン反応の速度定数k+2=0.46 s−1は,先行研究から得られた野生型酵素のk+2=2.2 s−1と比較して数倍低いながらも,H159Aにおいてパルミトイル-CoAのないときに進行するabortive transaminationの速度定数(4.4×10−4 s−1)よりもはるかに大きい値であった.この結果は,H159Aへのパルミトイル-CoAの結合によって,外アルジミンのCα–H結合の配向がイミン–ピリジン平面に対して垂直になるように固定されて脱プロトン反応が加速したが,Cα–H結合が脱プロトン反応に理想的な配向からわずかにずれているために野生型酵素の反応速度には及ばなかったと解釈される.k+2の比較から判断すると,H159Aではキノノイド中間体IIIが野生型酵素よりできにくいはずだが,実際には野生型酵素ではなくH159Aの反応系でIIIが観測された.この現象はグローバル解析の結果から以下のように説明される.クライゼン型縮合反応(図3, III→IV)の速度定数(k+3)はH159Aにおいては4.1 s−1と算出され,野生型酵素のk+3>75 s−1の20分の1近くに低下していた.つまり,キノノイドの生成速度の低下以上にキノノイドを減少させる反応の速度が低下したために,H159Aにおいてキノノイド中間体IIIが蓄積したのである.
表2 遷移状態下の反応解析から得られた反応動力学定数| 基質 | L-セリン | [α-2H]-L-セリン |
|---|
| Kd/mM | 0.0604±0.0001 | 0.0924±0.0002 |
| k+2/s−1 | 0.46±0.20 | 0.064±0.004 |
| k−2/s−1 | 3.48±2.26 | 2.19±0.49 |
| k+3/s−1 | 4.10±2.42 | 3.50±0.26 |
| k+4/s−1 | 3.30±0.12 | 3.34±0.19 |
| k−4/mM−1 s−1 | 262±21 | 140±37 |
7. クライゼン型縮合反応における酸触媒としてのHis159の役割
クライゼン型縮合の過程とはキノノイド中間体IIIのCαがカルボアニオンとしてパルミトイル-CoAのチオエステルを求核攻撃して新しい炭素−炭素結合を作ることである.カルボアニオンによる求核反応が進行するためにはパルミトイル-CoAのカルボニル酸素はプロトン化されることが望ましい.我々は野生型SPTの結晶構造に基づいた三者複合体構造モデル構築の結果からHis159のNε2はプロトン化されてパルミトイル-CoAのカルボニル酸素と水素結合を形成していると予想している8).求核攻撃に際してHis159が解離可能なNε2のプロトンをカルボニル酸素に供与することによってパルミトイル-CoAのチオエステル結合を活性化する可能性が高いと考えられる.つまり,His159は縮合反応のための一般酸触媒として作用する.
His159が芳香族アミノ酸残基に置換された変異型SPTにおいては,芳香環の水素原子ではパルミトイル-CoAのカルボニル酸素と水素結合を形成できず,さらに嵩高い芳香環がパルミトイル-CoAの適切な配置を妨げると考えられる.これがH159Aが酵素活性を保持できた一方でH159F,H159Y,およびH159Wが酵素活性を失った理由であろう.H159FとL-セリンとパルミトイル-CoAとの反応においてキノノイド中間体が観測されなかった実験結果とも矛盾しない.
時間分解スペクトルのグローバル解析の結果から算出されたクライゼン型縮合反応以降の過程の反応速度定数(k+4)はH159Aにおいて3.3 s−1と算出され,野生型酵素に比べてかなり低下していた.図5dはグローバル解析によって速度定数とともに算出される各中間体濃度の時間変化を表したものである.キノノイド成分としては,反応初期においてはPLP–L-セリンアルジミン中間体の脱プロトン化によって生成するキノノイド中間体IIIが主成分として蓄積するが,後期には縮合反応生成物が脱炭酸反応を経て生成するキノノイド中間体Vbが優勢となる.縮合反応以降の過程にHis159が影響しなければVb濃度はIIIと同様一定値に達し,このような曲線は得られないはずである.Vbの蓄積は,His159が縮合反応生成物からKDSへの変換過程をも促進している可能性を示唆している.
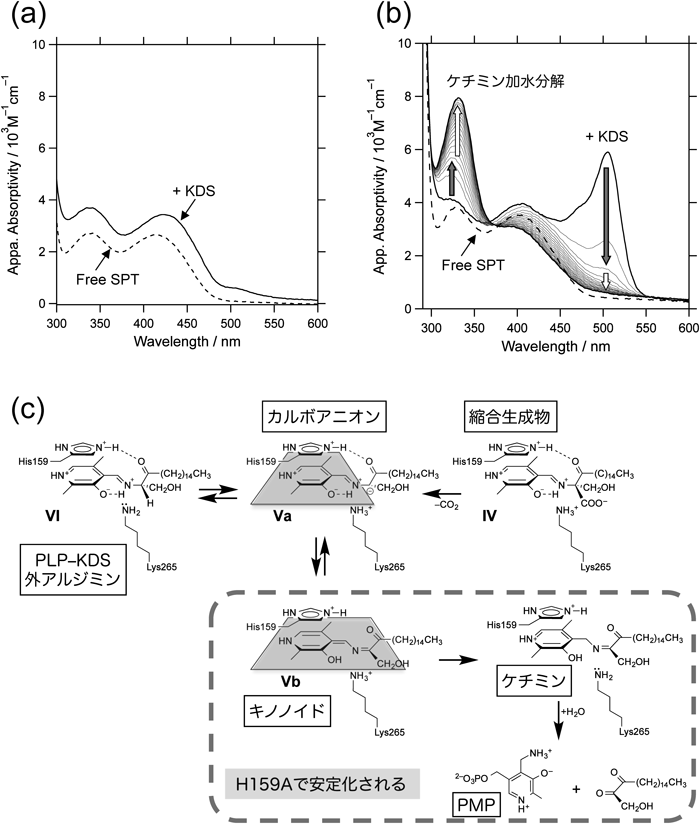
これに関する考察を可能にする結果が逆反応に該当するH159AとKDSの結合反応の分光学的解析から得られた.H159AにKDSを添加すると,野生型酵素ではほとんど検出されなかった新しい吸収ピークが505 nmに現れ,キノノイド分子種の蓄積が強く示唆された.この吸収スペクトルは形状,モル吸光係数ともに式1において想定した反応中間体D(キノノイド中間体Vb)と非常によく一致した(図6a, b;図5c).これらの結果は,観測されたのは,図6cに示すようにPLP–KDS外アルジミンの脱プロトンされた分子種であり,式1において想定した中間体Dは縮合生成物IVの脱炭酸によって生じるカルボアニオンVaと平衡状態にあるキノノイド中間体Vbであることを支持している.
H159AとKDSの反応によって生じたキノノイド中間体のスペクトルは不安定で,505 nmの吸収強度は次第に減少し,代わって330 nm付近の吸収が増加した(図6b).この吸収増加は二相性を示し,速い相は505 nmの吸収減少に対応した(図6b;灰色矢印).この現象は,比較的速いキノノイドのプロトン化によるケチミン中間体生成と,それに続くPMPとケトンへの加水分解(図6b;白色矢印)と解釈された(図6c).
想定されるキノノイド中間体Vbの構造はピリジン環からKDS由来部分のカルボニル炭素にまで伸びる平面構造をとる.KDSはカルボキシ基を持たないので,野生型SPTとKDSの複合体においては,His159とKDSのカルボニル酸素の間に水素結合が形成される可能性があるが,この水素結合はキノノイドの構造を歪めて不安定化すると推測される.このため,野生型SPTにおいてはカルボアニオンの状態をとりやすいはずである.一方,H159AではKDSカルボニル基とヒスチジン側鎖との相互作用がないためにキノノイド中間体が安定化されて蓄積したと考えられる.
野生型酵素ではKDSを出発基質とするabortive transamination(PLPのPMP化)が観測されず,一方でH159Aにおいてそれが観測されたこともこの推測を裏づけている(図6a, b).abortive transaminationが進行するにはKDSに由来する部分がケチミンという平面構造をとる必要があるが,His159との水素結合はこの平面構造を歪めてabortive transaminationを抑制するように作用する.野生型酵素においては,His159 Nε2とKDSカルボニル基の水素結合によってカルボアニオンのコンホメーションが固定され,そのプロトン化によるPLP−KDSアルジミン生成(最終生成物が酵素から解離して触媒サイクルが完結することへつながる)が促進されると考えられる.反応生成物KDSからのabortive transamination(好ましくない副反応)を抑制する点で,野生型酵素におけるカルボアニオンの構造は重要である.
また,パルミトイル-CoAやKDSのカルボニル基とHis159の相互作用を考えると,同様のカルボニル基とHis159間の相互作用が脱炭酸前の縮合生成物中間体においても形成されると推測される.脱炭酸反応がイミン–ピリジン共役系によって触媒されるためにはCα–COO−結合がイミン–ピリジン平面に対して垂直に配向することが必要となるが,この中間体においてはCα–COO−結合がイミン–ピリジン平面からのずれは30°にすぎず,むしろカルボニル基の平面に対して垂直になる.したがって,この結合はカルボニル基によって切断されると考えるのが妥当であり,His159がカルボニル基の酸素にプロトンを供与して脱炭酸に寄与すると予想される.つまり,His159は脱炭酸のステップにおいても酸触媒として作用すると考えられる.SPT阻害剤として知られるミリオシンはC2位にカルボキシ基,C3位にヒドロキシ基を有するKDS類似の化合物である.近年,野生型SPTとミリオシンの複合体結晶の構造解析の結果が報告された17).結晶中のミリオシンは脱炭酸された上に,ミリオシンの3位ヒドロキシ基とHis159の間に水素結合を形成しており,上述の反応機構を構造上から裏づける結果であった.
以上の実験結果に基づき,Sphingomonas SPTの反応制御機構についてまとめる(図3).L-セリンがSPTに結合するとSchiff塩基交換反応を経て外アルジミン中間体が生じる.SPTの外アルジミン中間体では,他の一般的なPLP酵素とは異なり,His159によってL-セリンのカルボキシ基が固定される.それにより,α位水素の配向がPLPピリジン環とSchiff塩基が作る平面に対して垂直になれず,結果として中間体の脱プロトン化による副反応が抑制されている.パルミトイル-CoAが結合すると,His159との水素結合の組換えによってL-セリンのコンホメーションが変化し,近傍のLys265によりα-プロトンが引き抜かれてキノノイド中間体が生じる.パルミトイル-CoAの有無による外アルジミン中間体のα-脱プロトン化の制御は,反応性の高いキノノイド中間体をむだに作らない点で合目的的である.縮合反応,CoAの解離と脱炭酸反応が進行してKDSが生じ,これが酵素から解離すると分子内Schiff塩基が再生する.全体を通して重要な役割を果たすのがHis159である.His159は,活性部位における基質の配向だけでなく生成物の配向も酵素反応の進行に合わせて厳密に制御し,両者からの副反応の進行を抑制する.また,His159は一般酸触媒としても働き,炭素–炭素結合の形成および脱炭酸を促進している.一つのアミノ酸残基による反応制御として実に絶妙である.ところが,これらは非律速段階であるために,従来の部位特異的変異解析では「変異による活性消失が認められないことから触媒性残基ではない」とされ,His159の真の役割が理解されない.しかし,SPT特異的な反応の進行を厳密に制御するためには,His159の多機能的役割は不可欠である.酵素反応の真の制御機構を理解するためには,触媒性残基のみならず反応特異性をつかさどる残基を見いだして解析することが重要であると思われる.
SPTはPLP酵素のα-オキサミン合成酵素(α-oxamine synthase: POAS)サブファミリーに分類され,これには5-アミノレブリン酸合成酵素(5-aminolevulinate synthase),8-アミノ-7-オキソノナン酸合成酵素(8-amino-7-oxononanoate synthase),2-アミノ-3-ケトブタン酸CoAリガーゼ(2-amino-3-ketobutyrate CoA ligase)などが属している13).いずれもアミノ酸とアシルCoA間の脱炭酸を伴う縮合反応を触媒する.これらの酵素はすべて結晶化されて立体構造が決定され,どの酵素の活性部位においても,SPTのHis159に相当するヒスチジン残基がPLPにスタッキングするように配置されていることが判明している.活性部位のヒスチジン残基の多機能性に支えられた副反応抑制の仕組みは,POASサブファミリーのPLP酵素群において共通する酵素反応制御機構であろう.
謝辞Acknowledgments
本研究に協力いただいた共同研究者と,JSPS科研費18570114,21570149,25440036,および日本応用酵素協会酵素研究助成に対し,この場を借りて深謝する.
引用文献References
1) Futerman, A.H. & Hannun, Y.A. (2004) EMBO Rep., 5, 777–782.
2) Hanada, K. (2003) Biochim. Biophys. Acta, 1632, 16–30.
3) Ikushiro, H., Hayashi, H., & Kagamiyama, H.(2000) Biochemistry and Molecular Biology of Vitamin B6 and PQQ-dependent Proteins (Iriarte, A., Kagan, H.M., & Martinez-Carrion, M., Eds.), pp 251-254, Birkhauser Verlag, Basel, Switzerland.
4) Ikushiro, H., Hayashi, H., & Kagamiyama, H. (2001) J. Biol. Chem., 276, 18249–18256.
5) Ikushiro, H., Hayashi, H., & Kagamiyama, H. (2003) Biochim. Biophys. Acta, 1647, 116–120.
6) Ikushiro, H., Islam, M.M., Tojo, H., & Hayashi, H. (2007) J. Bacteriol., 189, 5749–5761.
7) Ikushiro, H., Hayashi, H., & Kagamiyama, H. (2004) Biochemistry, 43, 1082–1092.
8) Ikushiro, H., Fujii, S., Shiraiwa, Y., & Hayashi, H. (2008) J. Biol. Chem., 283, 7542–7553.
9) Ikushiro, H., Okamoto, A., & Hayashi, H.(2006) Sphingolipid Biology (Hirabayashi, Y., Igarashi, Y., & Merrill, A., Eds.) pp. 483–492, Springer-Verlag Tokyo/Japan
10) Ikushiro, H., Islam, M.M., Okamoto, A., Hoseki, J., Murakawa, T., Fujii, S., Miyahara, I., & Hayashi, H. (2009) J. Biochem., 146, 549–562.
11) Shiraiwa, Y., Ikushiro, H., & Hayashi, H. (2009) J. Biol. Chem., 284, 15487–15495.
12) Ikushiro, H. & Hayashi, H. (2011) Biochim. Biophys. Acta, 1814, 1474–1480.
13) Schneider, G., Kack, H., & Lindqvist, Y. (2000) Structure, 8, R1–R6.
14) Yard, B.A., Carter, L.G., Johnson, K.A., Overton, I.M., Dorward, M., Liu, H., McMahon, S.A., Oke, M., Puech, D., Barton, G.J., Naismith, J.H., & Campopiano, D.J. (2007) J. Mol. Biol., 370, 870–886.
15) Raman, M.C., Johnson, K.A., Yard, B.A., Lowther, J., Carter, L.G., Naismith, J.H., & Campopiano, D.J. (2009) J. Biol. Chem., 284, 17328–17339.
16) Onuffer, J.J. & Kirsch, J.F. (1994) Protein Eng., 7, 413–424.
17) Wadsworth, J.M., Clarke, D.J., McMahon, S.A., Lowther, J.P., Beattie, A.E., Langridge-Smith, P.R., Broughton, H.B., Dunn, T.M., Naismith, J.H., & Campopiano, D.J. (2013) J. Am. Chem. Soc., 135, 14276–14285.
著者紹介Author Profile
 生城 浩子(いくしろ ひろこ)
生城 浩子(いくしろ ひろこ)大阪医科大学医学部生化学教室講師.博士(学術).
略歴1967年石川県に生る.90年金沢大学理学部化学科卒業.92年広島大学大学院生物圏科学研究科生物機能科学専攻博士課程(前期)修了.95年同大学院博士課程(後期)修了.同年(財)大阪バイオサイエンス研究所特別研究員.96年大阪医科大学医学部生化学教室(旧医化学教室)助手.2007年より現職.
研究テーマと抱負スフィンゴ脂質代謝に関わる酵素の構造と機能の解明.学生時代より一貫して酵素に関わっており,精製から反応機構の解析,立体構造決定をおこなってきた.生命現象の基盤となる酵素の働きを分子レベルで明らかにし,生命科学や医学研究に貢献することを目指している.
ウェブサイトhttp://www.osaka-med.ac.jp/deps/med/staff/ikushiro/
趣味水槽の掃除.