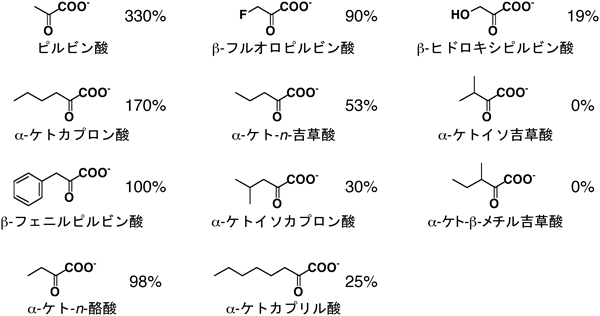
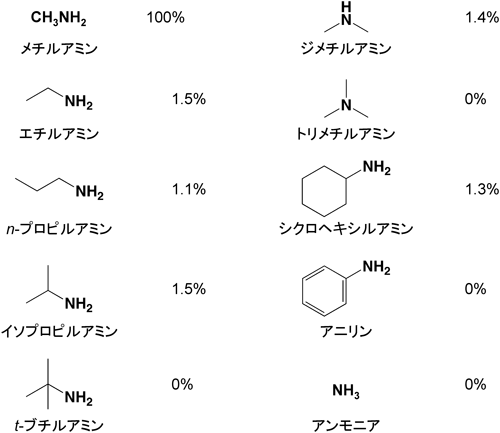
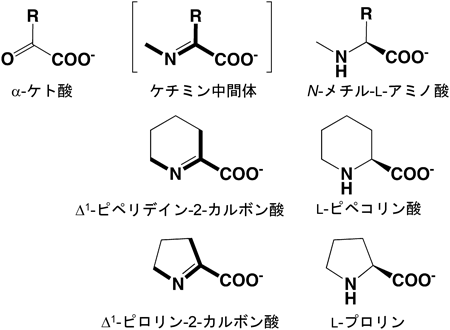
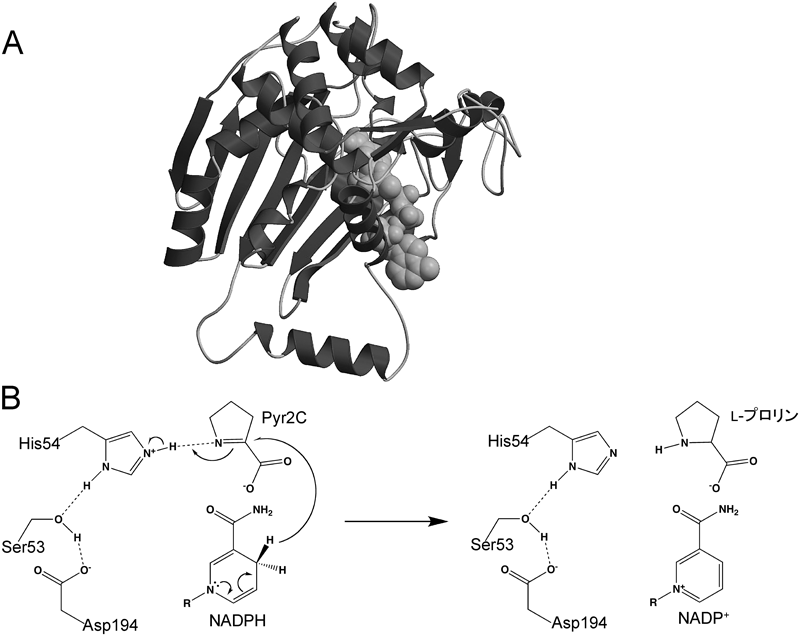
1) Pettit, G.R., Kamano, Y., Herald, C.L., Fujii, Y., Kizu, H., Boyd, M.R., Boettner, F.E., Doubek, D.L., Schmidt, J.M., Chapuis, J.C., & Michel, C. (1993) Tetrahedron, 49, 9151–9170.
6) Shaw, W.V., Tsai, L., & Stadtman, E.R. (1966) J. Biol. Chem., 241, 935–945.
7) Pollock, R.J. & Hersh, L.B. (1971) J. Biol. Chem., 246, 4737–4743.
8) Latypova, E., Yang, S., Wang, Y.S., Wang, T.S., Chavkin, T.A., Hackett, M., Schafer, H., & Kalyuzhnaya, M.G. (2010) Mol. Microbiol., 75, 426–439.
9) Lin, M.C.M. & Wagner, C. (1975) J. Biol. Chem., 250, 3746–3751.
15) Nelson, K.E., Weinel, C., Paulsen, I.T., Dodson, R.J., Hilbert, H., dos Santos, V.A.P.M., Fouts, D.E., Gill, S.R., Pop, M., Holmes, M., Brinkac, L., Beanan, M., DeBoy, R.T., Daugherty, S., Kolonay, J., Madupu, R., Nelson, W., White, O., Peterson, J., Khouri, H., Hance, I., Lee, P.C., Holtzapple, E., Scanlan, D., Tran, K., Moazzez, A., Utterback, T., Rizzo, M., Lee, K., Kosack, D., Moestl, D., Wedler, H., Lauber, J., Stjepandic, D., Hoheisel, J., Straetz, M., Heim, S., Kiewitz, C., Eisen, J., Timmis, K.N., Dusterhoft, A., Tummler, B., & Fraser, C.M. (2002) Environ. Microbiol., 4, 799–808.
20) Forouhar, F., Lee, I., Benach, J., Kulkarni, K., Xiao, R., Acton, T.B., Montelione, G.T., & Tong, L. (2004) J. Biol. Chem., 279, 13148–13155.
23) Chang, Y.F. & Adams, E. (1974) J. Bacteriol., 117, 753–764.
24) Payton, C.W. & Chang, Y.F. (1982) J. Bacteriol., 149, 864–871.
25) Meister, A., Radhakrishnan, A.N., & Buckley, S.D. (1957) J. Biol. Chem., 229, 789–800.
26) Goto, M., Muramatsu, H., Mihara, H., Kurihara, T., Esaki, N., Omi, R., Miyahara, I., & Hirotsu, K. (2005) J. Biol. Chem., 280, 40875–40884.
28) Tanaka, H., Kuroda, A., Marusawa, H., Hatanaka, H., Kino, T., Goto, T., Hashimoto, M., & Taga, T. (1987) J. Am. Chem. Soc., 109, 5031–5033.
29) Germann, U.A., Shlyakhter, D., Mason, V.S., Zelle, R.E., Duffy, J.P., Galullo, V., Armistead, D.M., Saunders, J.O., Boger, J., & Harding, M.W. (1997) Anticancer Drugs, 8, 125–140.