2) Haynes, L. & Lefebvre, J.S. (2011) Aging Dis., 2, 374–381.
3) Shaw, A.C., Joshi, S., Greenwood, H., Panda, A., & Lord, J.M. (2010) Curr. Opin. Immunol., 22, 507–513.
4) Maue, A.C., Yager, E.J., Swain, S.L., Woodland, D.L., Blackman, M.A., & Haynes, L. (2009) Trends Immunol., 30, 301–305.
6) Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., & Kirkland, J.L. (2013) J. Clin. Invest., 123, 966–972.
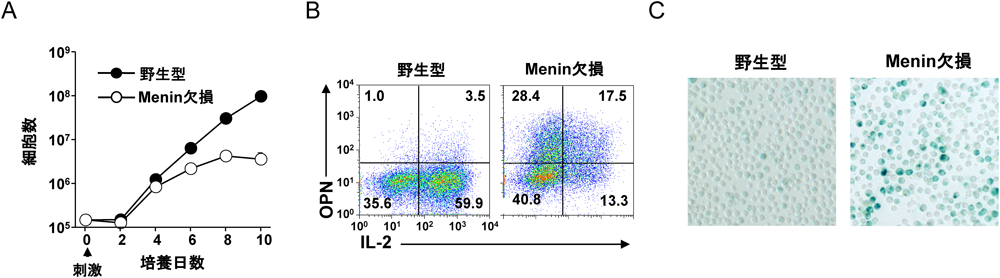
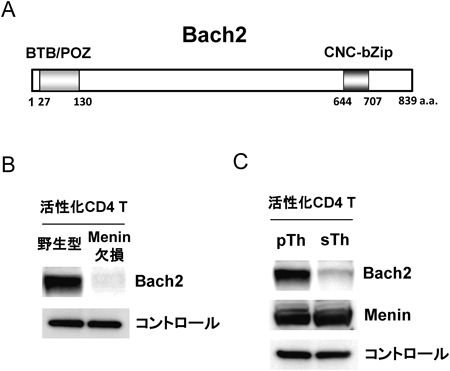
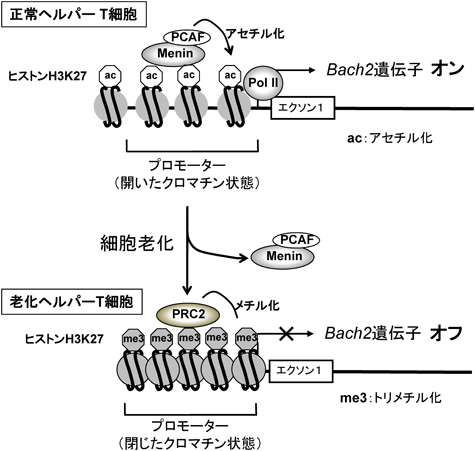
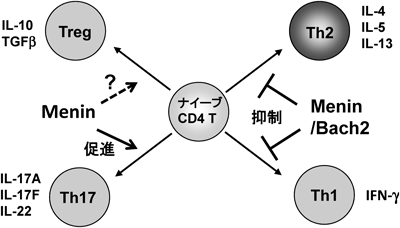
7) Kuwahara, M., Suzuki, J., Tofukuji, S., Yamada, T., Kanoh, M., Matsumoto, A., Maruyama, S., Kometani, K., Kurosaki, T., Ohara, O., Nakayama, T., & Yamashita, M. (2014) Nat. Commun., 5, 3555.
9) Yamashita, M., Hirahara, K., Shinnakasu, R., Hosokawa, H., Norikane, S., Kimura, M.Y., Hasegawa, A., & Nakayama, T. (2006) Immunity, 24, 611–622.
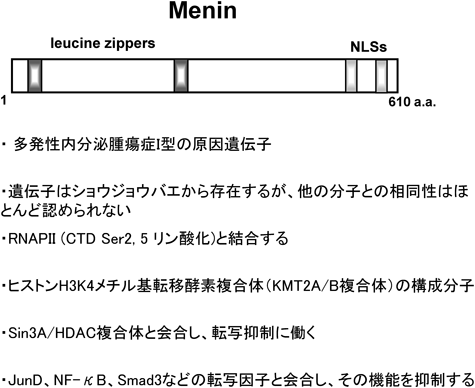
10) Brandi, M.L., Gagel, R.F., Angeli, A., Bilezikian, J.P., Beck-Peccoz, P., Bordi, C., Conte-Devolx, B., Falchetti, A., Gheri, R.G., Libroia, A., Lips, C.J., Lombardi, G., Mannelli, M., Pacini, F., Ponder, B.A., Raue, F., Skogseid, B., Tamburrano, G., Thakker, R.V., Thompson, N.W., Tomassetti, P., Tonelli, F., Wells, S.A. Jr., & Marx, S.J. (2001) J. Clin. Rndocrinol. Metab., 86, 5658–5671.
13) Shimatani, K., Nakashima, Y., Hattori, M., Hamazaki, Y., & Minato, N. (2009) Proc. Natl. Acad. Sci. USA, 106, 15807–15812.
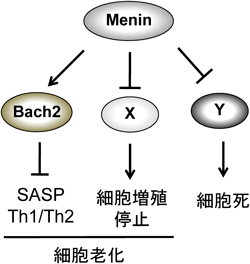
14) Muto, A., Ochiai, K., Kimura, Y., Itoh-Nakadai, A., Calame, K.L., Ikebe, D., Tashiro, S., & Igarashi, K. (2010) EMBO J., 29, 4048–4061.
15) Muto, A., Tashiro, S., Nakajima, O., Hoshino, H., Takahashi, S., Sakoda, E., Ikebe, D., Yamamoto, M., & Igarashi, K. (2004) Nature, 429, 566–571.
16) Kometani, K., Nakagawa, R., Shinnakasu, R., Kaji, T., Rybouchkin, A., Moriyama, S., Ikebe, D., Yamamoto, M., & Igarashi, K. (2013) Immunity, 39, 136–147.
17) Itoh-Nakadai, A., Hikota, R., Muto, A., Kometani, K., Watanabe-Matsui, M., Sato, Y., Kobayashi, M., Nakamura, A., Miura, Y., Yano, Y., Tashiro, S., Sun, J., Ikawa, T., Ochiai, K., Kurosaki, T., & Igarashi, K. (2014) Nat. Immunol., 15, 1171–1180.
18) Roychoudhuri, R., Hirahara, K., Mousavi, K., Clever, D., Klebanoff, C.A., Bonelli, M., Sciumè, G., Zare, H., Vahedi, G., Dema, B., Yu, Z., Liu, H., Takahashi, H., Rao, M., Muranski, P., Crompton, J.G., Punkosdy, G., Bedognetti, D., Wang, E., Hoffmann, V., Rivera, J., Marincola, F.M., Nakamura, A., Sartorelli, V., Kanno, Y., Gattinoni, L., Muto, A., Igarashi, K., O’Shea, J.J., & Restifo, N.P. (2013) Nature, 498, 506–510.
19) Dohi, Y., Ikura, T., Hoshikawa, Y., Katoh, Y., Ota, K., Nakanome, A., Muto, A., Omura, S., Ohta, T., Ito, A., Yoshida, M., Noda, T., & Igarashi, K. (2008) Nat. Struct. Mol. Biol., 15, 1246–1254.
20) Lee, W.W., Kang, S.W., Choi, J., Lee, S.H., Shah, K., Eynon, E.E., Flavell, R.A., & Kang, I. (2010) Blood, 115, 530–540.
21) Tsukumo, S., Unno, M., Muto, A., Takeuchi, A., Kometani, K., Kurosaki, T., Igarashi, K., & Saito, T. (2013) Proc. Natl. Acad. Sci. USA, 110, 10735–10740.
22) Watanabe, Y., Onodera, A., Kanai, U., Ichikawa, T., Obata-Ninomiya, K., Wada, T., Kiuchi, M., Iwamura, C., Tumes, D.J., Shinoda, K., Yagi, R., Motohashi, S., Hirahara, K., & Nakayama, T. (2014) Proc. Natl. Acad. Sci. USA, 111, 12829–12834.
23) Mannick, J.B., Del Giudice, G., Lattanzi, M., Valiante, N.M., Praestgaard, J., Huang, B., Lonetto, M.A., Maecker, H.T., Kovarik, J., Carson, S., Glass, D.J., & Klickstein, L.B. (2014) Sci. Transl. Med., 6, 268ra179.