我々を取りまく環境には,人体に有害なウイルス・微生物など多種多様の異物が存在しており,これらから身を守るために,人体は“免疫”という生体防御機構を備えている.しかし,この免疫の礎となる機構「免疫寛容(トレランス)」が“生まれながらにして”機能していないのが「自己免疫疾患」という難病であり,現在のところ,根治療法が確立されていない.自己免疫疾患は自己を“守る”はずの機構が自己を“攻撃”してしまう希有な疾患であり,全身性エリテマトーデス(systemic lupus erythematosus: SLE)や慢性関節リウマチ(rheumatoid Arthritis: RA)など,40種類あまりが報告されている.これら自己免疫疾患の原因には,遺伝因子や環境因子などが複雑に関与しているため,いまだに,その発症機構はよくわかっていない.しかし,多くの自己免疫疾患のなかで,家系分析等の結果から,単一遺伝子の突然変異によって発症することがわかっているきわめてまれな自己免疫疾患(単一遺伝子病)が2種類知られている.それらは,自己免疫性多腺性内分泌不全症(autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy: APECED, OMIM #240300)とX染色体連鎖型免疫調節異常(immunodysregulation-polyendocrinopathy-enteropathy-X-linked syndrome: IPEX, OMIM #304790)である.
我々は,主にフィンランドの患者DNAを用いてAPECEDの原因遺伝子のクローニングに成功し,AIRE(エア:autoimmune regulator)と名づけた1).一方,IPEXの原因遺伝子はFOXP3(Forkhead box P3)と同定された2).AIRE遺伝子は健常者では主に胸腺の髄質領域の希少な上皮細胞で発現しており,中枢性の免疫寛容に関与している.また,FOXP3は末梢の制御性T細胞で発現し,末梢性の免疫寛容に関与している.
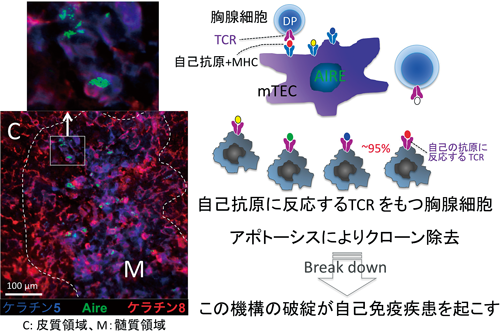
中枢性免疫寛容の成立をつかさどる臓器である胸腺は,皮質と髄質からなるが,それらを構成する細胞の90%強が胸腺細胞(thymocyte)であり,細胞表面抗原であるCD4やCD8を発現しているか否かにより大きくDN(double negative),DP(double positive),SP(single positive: CD4もしくはCD8どちらか一方が陽性)の分化段階に分けられる.皮質領域の最も未成熟なDNは,皮質–髄質境界領域へ移動するとDPとなり,自己反応性を示すものが除去され,自己の抗原に反応しない胸腺細胞がSPへと分化し,髄質領域から末梢血へと放出される.残り10%弱が上皮細胞(ストロマ細胞:stroma cell)すなわち胸腺皮質上皮細胞(cortical thymic epithelial cell: cTEC)と胸腺髄質上皮細胞(medullary thymic epithelial cell: mTEC)である.さらに,胸腺には樹状細胞(dendritic cell: DC)が存在する.胸腺上皮細胞は胸腺内で網目状構造を作り,その間隙を移動する胸腺細胞のうち“自己にとって不利”となる胸腺細胞を除去している.実に,胸腺細胞の約95%が自己にとって不利なものであり,除去は二つの機構によって行われている.すなわち,①皮質領域の上皮細胞が行う「正の選択(ポジティブセレクション)」と,②髄質領域の上皮細胞が行う「負の選択(ネガティブセレクション)」である(図1).特に,胸腺におけるネガティブセレクションではアポトーシスによる自己反応性T細胞のクローン除去が行われている(図2,unpublished data).その結果,わずか5%程度の胸腺細胞が末梢に放出され,それらがやがて感染防御を担うT細胞となる.
T細胞のポジティブセレクションは,胸腺細胞上のT細胞受容体(T cell receptor: TCR)が自己の主要組織適合遺伝子複合体(major histocompatibility complex: MHC)を適切に認識するかをチェックする機構であり,T細胞のネガティブセレクションは,胸腺細胞上のTCRと自己抗原・自己MHCの結合強度をチェックする機構である.これら二つのチェック機構により,自己のMHCを認識しない,もしくは自己抗原と弱くまたは強く反応する胸腺細胞が除去される.
AIRE遺伝子の発見(1997年)から17年あまり経過したが,副甲状腺機能低下症や副腎皮質機能低下症,1型糖尿病などを併発した患者は,幼少期における皮膚粘膜カンジダ症など症状の組み合わせからAPECEDが疑われ,その最終診断であるAIRE遺伝子の変異により確定される.日本人におけるAPECED患者も2002年に同定されている3).また,世界中で臨床・基礎両面での研究成果が次々と公表され,その発症機構の理解も少しずつ進んでいる.
本稿では,まず自己免疫疾患APECEDの臨床症状を外観し,次いで我々がクローニングしたAIRE遺伝子とそれから作られるAIREタンパク質の特徴を述べる.さらに,胸腺の髄質領域に存在する希有な上皮細胞,すなわち,マウスAire遺伝子を発現している(Aire+)細胞を三つ(Aire+TEC1, Aire+TEC2, Aire+DC)株化した経過を述べる.また,それらAire+細胞株と新鮮胸腺細胞との共培養システムを確立して,“T細胞のネガティブセレクション”をin vitroで再現できたことを紹介する.さらに,このin vitroシステムを用いて,“免疫寛容”の成立や“胸腺クロストーク”の分子機構に関して,新たな情報が得られつつある状況を述べる.今後,三つのAire+細胞株は,自己免疫疾患(APECEDなど)の発症機構を胸腺髄質上皮細胞からの視点で追究するための有益なツールになると期待している.
自己免疫疾患は,生体防御の礎となる「免疫寛容」に異常が生じ,自己のT細胞が自己の正常細胞に反応したり,自己抗体が産生されたりすることによって発症する.SLEやRA, 1型糖尿病(type I diabetes),多発性硬化症(multiple sclerosis),や炎症性腸疾患(inflammatory bowel disease)など多くの自己免疫疾患は,その病因が多因子によるため,発症メカニズムの解明はきわめて困難である.
一方,単一遺伝子病APECEDの患者では,副腎皮質機能低下症(アジソン病),副甲状腺機能低下症,皮膚粘膜カンジダ症などの3主症状が高率に合併している.さらに,性腺機能低下症,脱毛,吸収不全症,白斑,慢性肝炎,角膜症なども併発している.このように,APECEDは単一遺伝子病であるにもかかわらず,その臨床症状はきわめて多彩である.
アジソン病や性腺機能低下症を示すAPECED患者の血清には抗P450scc,抗P450c17,抗P450c21などの自己抗体が,糖尿病を併発するAPECED患者の血清中にはIA2β,インスリン,プロインスリン,GAD2(glutamate decarboxylase 2)などの自己抗原に対する自己抗体が検出される(表1).つまり,APECED患者では,臓器に特異的なタンパク質に対する“自己抗体や自己反応性T細胞”が産生されることで機能障害が生じている.
表1 APECED患者の臨床症状の特徴| 臨床所見 | 自己抗原タンパク質・遺伝子 | 組織特異性 |
|---|
| 内分泌系 | 上皮小体不全症 | CASR (calcium sensing receptor) | 甲状腺 |
| アジソン病 | CYP21 | 副腎 |
| CYP17a | 生殖腺 |
| 生殖腺不全 | CYP11A | 生殖腺 |
| 甲状腺機能低下症 | TPO (tyroid peroxydase) | 甲状腺 |
| TGN (thyroglobulin) | 甲状腺 |
| インスリン依存糖尿病 | GAD2 (GAD65) | 膵臓 |
| PTPRN (IA-2) | 膵臓 |
| 内分泌系以外 | 白斑症 | SOX9 (SRY-box 9) | 色素細胞 |
| 脱毛症 | TH (tyrosine hydroxylase) | 脳 |
| 吸収不良症 | TPH (tryptophan hydroxylase) | 小腸 |
| 自己免疫肝炎 | CYP1A2 | |
| DDC (AADC) | 肝臓 |
3. APECEDの原因遺伝子のマッピングとクローニング
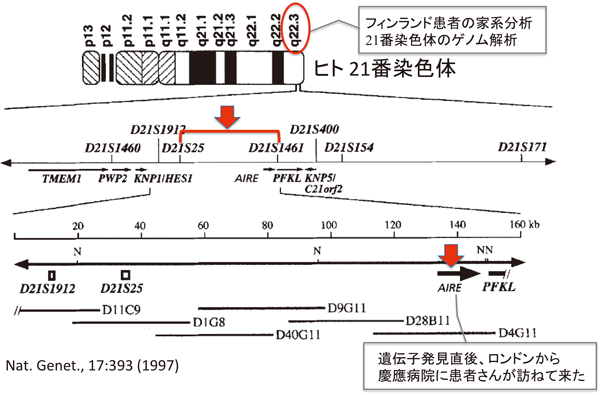
家系分析から,APECEDは単一遺伝子からなる常染色体劣性遺伝形式をとることが明らかにされた.健常人と患者末梢血由来のゲノムDNAサンプルを用いて多数のSTS(sequence tagged site)マーカーに関するPCR産物の断片長解析(haplotype mapping)を繰り返し行い,その遺伝子座位(locus)をヒト21番染色体のバンド22.3領域にマップした4,5).さらに,ポジショナルクローニングの手法により,21番染色体上のSTSマーカーを用いて,原因遺伝子の候補を160 kbのDNA領域に狭め,その中に位置する遺伝子の変異を逐次解析し,APECED患者に特有の突然変異を見つけ,原因遺伝子として確証を得(図3),その遺伝子をautoimmune regulator(AIRE)と命名した.
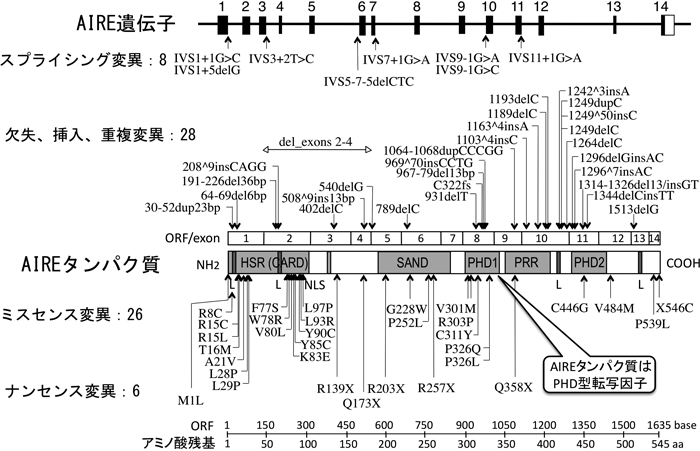
AIRE遺伝子は1997年に発見されてから現在までに,世界中の患者より,約70種類の異なる突然変異(スプライシング変異,欠失,挿入,重複変異,ミスセンス変異,ナンセンス変異)が遺伝子のあらゆる部分に見いだされている1,3,6)(図4上段).
4. APECEDの原因遺伝子が作るAIREタンパク質は転写因子であり,いくつかの調節タンパクと相互作用する
AIRE遺伝子のゲノムサイズは1.635 kbで,14個のエキソンからなり,2257 bpのmRNAが転写され,545アミノ酸残基からなる約60 kDaのAIREタンパク質が生合成(翻訳)される.AIREタンパク質の構造は,4個のLXXLLドメイン(核内受容体結合モチーフ),HSRドメイン(homogenously staining region,二量体形成に関与),プロリンリッチ領域,SANDドメイン(DNA結合能を有する),2個のPHDドメイン(plant homeodomain,非メチル化ヒストンとの結合能を持つ)からなる.含有するドメインの機能特性より,AIREタンパク質は「PHD型転写因子」と推定された(図4中央).
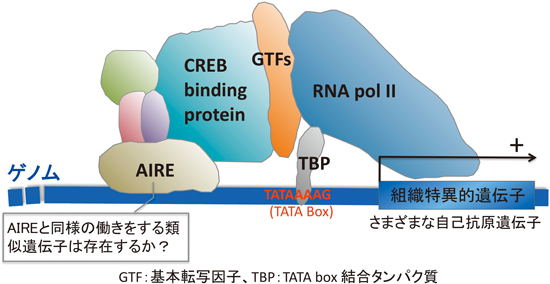
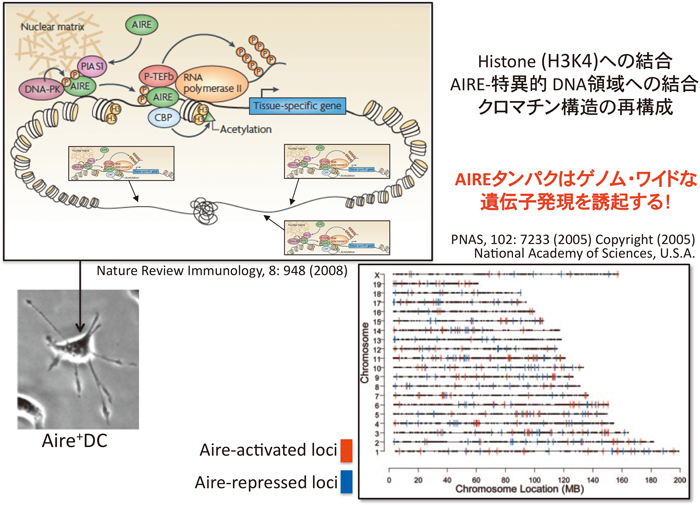
AIRE遺伝子のクローニングの直後に,それと結合するタンパク質として一番初めに同定されたものは,CREB(cyclic AMP-responsive element binding protein)-binding protein(CBP)であった7).これは,AIREが転写活性制御に関与することを初めて示唆したデータ(図5)であったので,転写コアクチベーターであるCBPと相互作用するAIREは,胸腺ネガティブセレクションを司る胸腺髄質上皮細胞内で発現する遺伝子群の転写制御を行っているものと考えた.
臓器・組織レベルでのAIRE遺伝子の発現は,胸腺における一部の髄質上皮細胞で最も強くみられ,次いでリンパ節での発現が高い.さらにより高感度な細胞レベルでの検出法では,肝臓,脾臓,末梢血中の単球や樹状細胞などでも,AIRE遺伝子の発現が確認されている.中枢性免疫寛容は胸腺において,末梢性免疫寛容はリンパ節や末梢血において,それらの機構が成立している.このように,中枢性/末梢性いずれにおいてもAIREは,免疫寛容機構に深く関連しており,1997年発見当初AIRE遺伝子は,“自己の免疫を調節する遺伝子”という思いで命名したのだが,まさに,T細胞が攻撃してはいけない臓器特異的なタンパク質(自己抗原)に関する遺伝子群の発現を転写因子として調節していることが次第にわかってきた.
また図4のようにAIREタンパク質分子には2個のPHDドメインが見いだされており,PHD1はE3ユビキチン活性を持ち,PHD2は転写活性に関与するドメインである7,8).特に,AIREタンパク質分子にユビキチン活性が推定されたことは興味深い.なぜなら,ヒストンのユビキチン化や転写コファクターの分解などは,RNAポリメラーゼIIの伸長反応を促進し,転写を調節しているといわれているからである9).
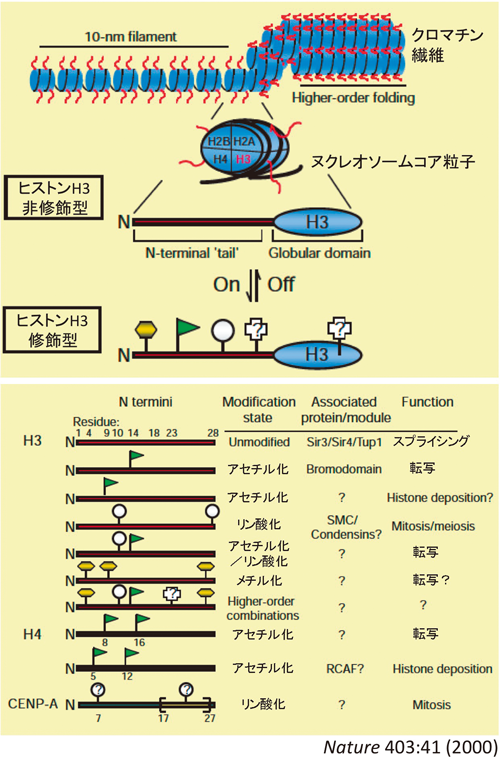
5. AIRE遺伝子の発現はヒストンの修飾状態によって,クロマチンレベルで制御されている
一般に,コアヒストンは,H2A,H2B,H3,H4という4種のヒストンタンパク質からなり,さらに,H2A–H2Bの二量体が2個と,H3–H4の四量体が1個集合した八量体として存在している.
ヒストンはメチル化,アセチル化,リン酸化,ユビキチン化,ADPリボシル化などの化学修飾を受け,その修飾の組み合わせにより,転写制御がなされている(図6,ヒストンコード仮説).一般に,ヒストンH3では,4番目と36番目のリシン残基のメチル化は転写を活性化させ,9番目,27番目,79番目のリシン残基のメチル化は逆に,転写を抑制させる.AIREの転写制御は,ヒストンH3の4番目(H3K4)のメチル化状態に依存している10–12).すなわちAIREタンパク質は,標的遺伝子のプロモーター領域における三重メチル化されたヒストンH3K4(H3K4me3)やアセチル化されたヒストンH3(AcH3)などで修飾されたクロマチンには結合しないため,転写を促進しない.逆に,低メチル化H3K4のクロマチン(H3K4me0)には結合して,転写活性を示す.
6. ノックアウト(Aire−/−)マウスの解析から確認されたこと
Aireノックアウト(Aire−/−)マウスは,2002年Peltonenらによって作られた.このAire−/−マウスでは,ヒトAPECED患者と同様,末梢組織へのリンパ球浸潤や自己抗体の産生によって,標的臓器の免疫障害を起こしていた.さらに,胸腺の髄質上皮細胞で末梢組織に特異的な遺伝子群(自己抗原)の発現が低下していた13).つまり,ヒトAPECED患者の病状を再現できるマウスモデルとして有用であることが示され,その後各方面で活用されている.2003年ListonらのAire−/−マウスにおいては,Aireの変異により膵臓ランゲルハンス島に対する自己反応性T細胞が除去されないことが証明され14),2005年JiangらのAire−/−マウスにおいては,NOD,B6,BALB/cなどマウスの種により自己抗体の標的臓器が異なることが明らかにされた15).これはAPECED患者において異なる臨床症状を呈していることと関係していると思われる.また,2005年黒田らのAire−/−マウスでは,胸腺上皮細胞におけるα-Fodrinやpancreas-specific protein disulfide isomerase(PDIp)のmRNAレベルに発現低下が認められないものの,両分子に対する自己抗体が認められた16).つまり転写活性が低下していない自己抗原に対して,自己寛容の破綻が認められたのである.これは,AIREが自己抗原の提示過程にも関与しているなど,転写活性以外にもネガティブセレクションに作用している可能性があることを示唆している.
7. AIREタンパク質は複数のタンパク質と相互作用し,多様な機能に関与している
ヒトHEK293T細胞や胸腺上皮細胞株にタグ融合型AIREタンパク質を強制発現させ,細胞抽出液を調製して共免疫沈降を行ったところ,AIREと相互作用するタンパク質が複数同定された.それらは推定される機能から次の2群に分類された.①群:DNA-PK(DNA-dependent protein kinase),PARP-1(poly-ADP ribose-polymerase family, member 1),TOP2a(topoisomerase 2a),FACT(facilitates chromatin transcription complex subunit),Ku 70 & Ku80(Ku protein 70 & 80),H2AX(histone H2AX protein).AIREタンパク質はこれらの核内タンパク質と相互作用することによって,複数の標的遺伝子の転写を制御していると推察された.②群:RNAヘリカーゼやスプライソソームの構成分子等.このことから,AIREタンパク質はpre-mRNAのプロセッシングにも関与していることが示唆された17).
8. Aire遺伝子を恒常的に発現する細胞株を樹立する
転写因子AIREの真の機能を解明するためには,本来,内在的(endogenous)かつ恒常的にAIREを発現している細胞を用いることが理想である.我々はマウス胸腺でAIREタンパク質をendogenousに発現している希有な髄質上皮細胞の株化を試み,実際に,三つの細胞株を樹立した18).
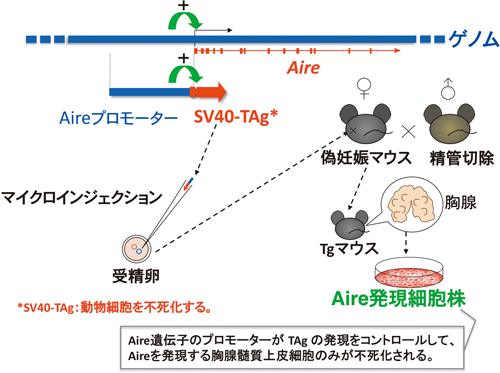
細胞株の樹立化に向けて,まず,マウス全ゲノムをカバーするファージDNAライブラリーをスクリーニングして,Aire遺伝子上流のプロモーター領域を含むファージクローンを単離した.次にAire遺伝子に隣接する遺伝子Dnmt3lまでの領域を含むクローンを得,それから,Aire遺伝子の開始コドンより上流約6.3 kbのゲノムDNA断片を切り出した.さらに,その断片の下流にSV40 Large T抗原(TAg)遺伝子(哺乳動物細胞を不死化させる特質を有する)を連結して,トランスジーン(Tg)を作製した(図7).
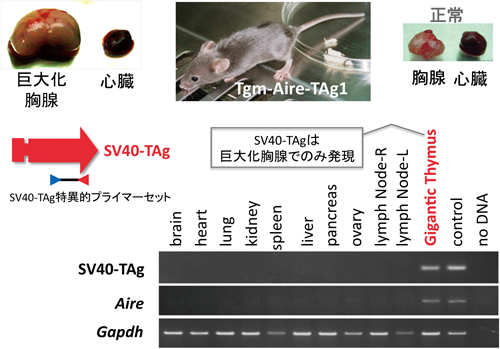
次いで,そのTgをマウス受精卵にマイクロインジェクションして,発生を続けさせ,Aireのプロモーター領域とSV40-TAgからなるTgを保持するトランスジェニックマウス(Tgn-Aire-TAg)を得た.そのTgn-Aire-TAgは生後40日目に呼吸困難に陥ったため開胸すると,胸腺が肥大化していた(図8).
この肥大(巨大)化胸腺とともに,9個の臓器(脳,心臓,肺,腎臓,脾臓,肝臓,膵臓,卵巣,リンパ節)を摘出し,各薄切切片の免疫組織化学染色を行ったところ,巨大化胸腺でのみSV40-TAgに陽性の上皮細胞が存在していた.この結果から,Aire-TAg融合遺伝子は,Aireが発現される胸腺の希有な細胞でのみ働き,T抗原が作られ細胞が不死化し,胸腺が肥大化したと考えた.
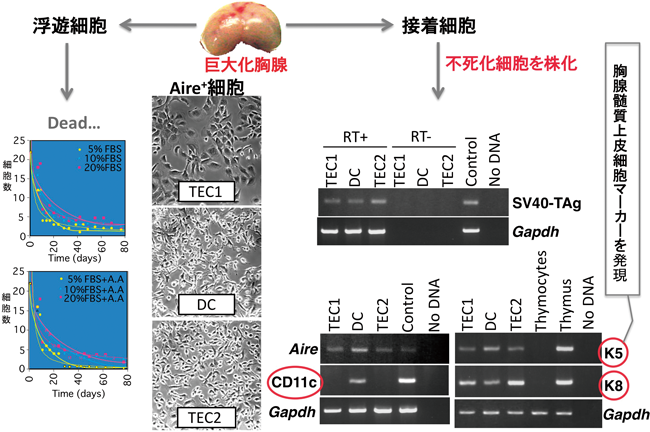
巨大化胸腺を解剖し,胸腺細胞と上皮細胞の画分を得た.胸腺細胞画分に対して必須アミノ酸を含むないし含まない培地に,5%,10%,20%のウシ胎仔血清を加えた計六つの条件下にて培養したところ,浮遊している胸腺細胞はいかなる培養条件下においても増殖できず,培養開始80日後には全部死滅した.一方,接着した上皮細胞の一部は無限増殖を示したので,コロニーを形成する細胞群をクローニングカップにて単離した.さらに,3回の限外希釈法にて単一細胞種からなる3種のAire+細胞株(Aire+TEC1, TEC2, DC)を樹立した(図9中央写真,右写真上段).三つのAire+細胞株は,いずれも,AireとSV40-T抗原をendogenousに発現していた.さらに,「ケラチン5とケラチン8(K5, K8)」を発現していた(図9右写真下段).つまり,いずれも胸腺の髄質皮質上皮細胞由来であった.そのうちの一つAire+DC株は,樹状細胞のマーカーであるCD11cも発現していた.
9. 三つのAIRE+細胞株はすべて自己抗原提示細胞の特性を持っていた
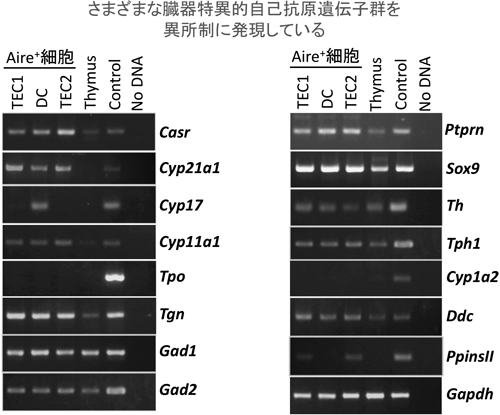
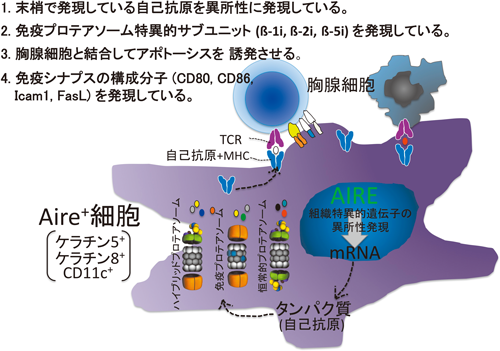
三つのAire+細胞株は,APECED患者血清中に存在する複数の自己抗体の標的となる自己抗原遺伝子,すなわち膵臓に特有のPTPRN(protein tyrosine phosphatase, receptor type, N)や肝臓に特有のCYP1A2(cytochrome P450, family 1, subfamily A, polypeptide 2)などを発現していた(図10).これらは,まさしく胸腺の自己抗原提示細胞(self-antigen presenting cell: self-APC)が示す「異所性発現」という特性である.
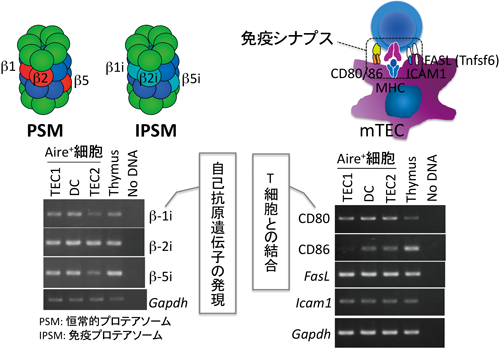
通常の細胞はプロテアソームでタンパク質のプロセッシングを行うが,胸腺APCにおいては恒常的プロテアソームに代わり,免疫プロテアソームが行っている.免疫プロテアソームは,恒常的プロテアソームのサブユニット(β1, β2, β5)を特異的なサブユニット(β1i, β2i, β5i)に置き換えて作られている.これらサブユニットの発現が,胸腺髄質上皮細胞における自己抗原プロセッシングに必須である.三つのAire+細胞株すべてで,それら免疫プロテアソーム特有のサブユニットが発現されていた(図11左側).
self-APCのさらなる特性は,T細胞と結合するための「免疫シナプス」を形成することである.このときのAPC側の構成成分として,CD80,CD86,FasL,Icam1が知られているが,これらは実際に,Aire+細胞で発現されていた(図11右側).このように,樹立した3種のAire+細胞株は,self-APCの3大特徴(末梢組織特有の遺伝子の発現,免疫プロテアソーム,免疫シナプス)をすべて保持していた.
ここまで株化されたAire+細胞の性状を述べたが,これら細胞株を用いて,新鮮胸腺細胞との共培養システムを確立した.まず,“T細胞のネガティブセレクション”をin vitroで再現できたことを紹介する.さらに,このin vitroシステムを用いて,“免疫寛容”の成立や“胸腺クロストーク”の分子機構に関して,新たな情報が得られつつある状況を述べる.
10. Aire+細胞株を用いて胸腺におけるネガティブセレクションをin vitroで再現する
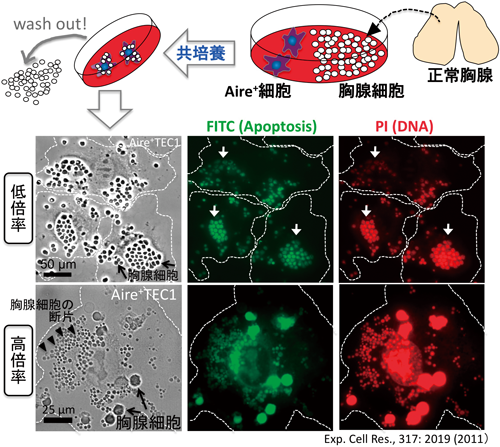
胸腺から得た新鮮な胸腺細胞群をAire+細胞株とin vitroで共培養すると,Aire+細胞は遊走しながら胸腺細胞を捕らえた(図12,動画1).
Aire+細胞に捕らえられた胸腺細胞は,PBSで洗浄してもはがれ落ちることはなく,これらの胸腺細胞はアポトーシスに陥っていることが観察された.つまり,in vitroで胸腺におけるネガティブセレクションを再現できた.
一方,正常な末梢血にも1~5%の自己反応性T細胞が存在することが知られている.実際に,Aire+細胞は末梢血リンパ球の一部と結合した.その数は,正常マウスからよりもAireノックアウトマウス(Aire依存性T細胞ネガティブセレクション機能を欠損)からの方が顕著に多かった.
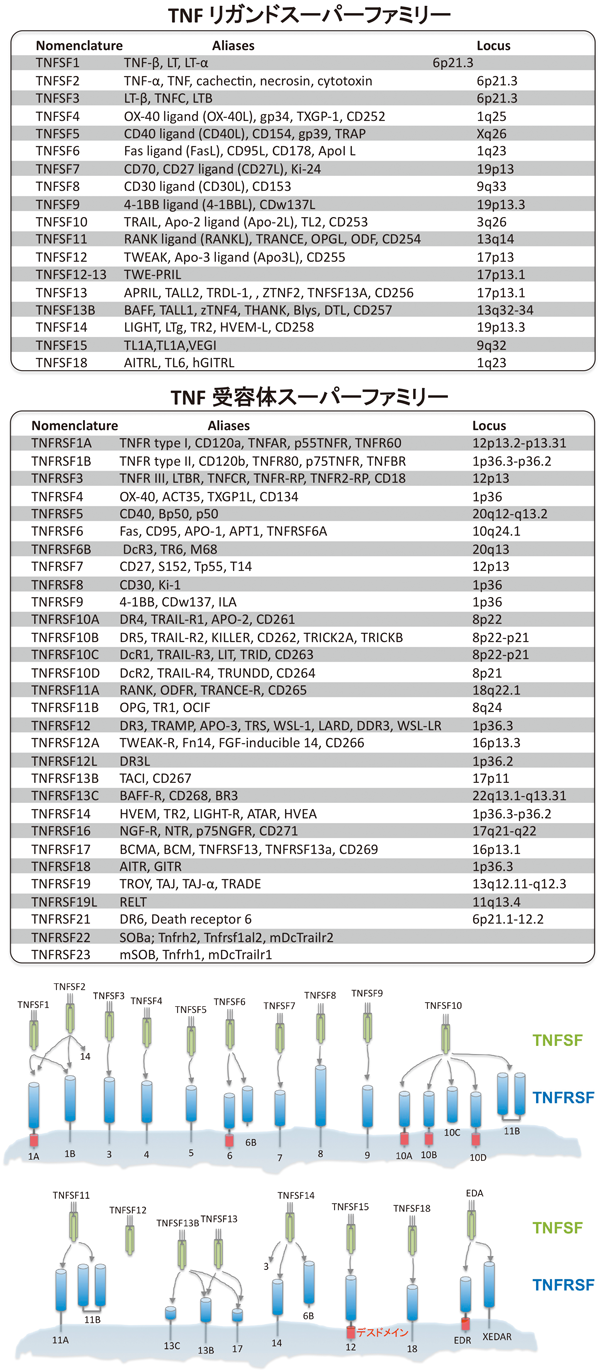
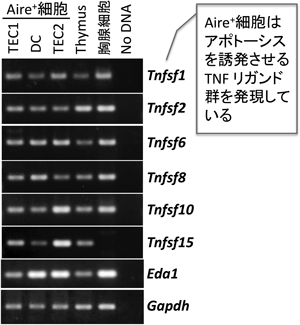
自己反応性胸腺細胞の除去機構について,1992年Demanらが「tumor necrosis factor(TNF)-αが胸腺で発現している」ことを発見したが,これを皮切りに“胸腺におけるTNFの役割”が研究されている.現在,TNFは18種類のTNFリガンドスーパーファミリー(TNFSF)と30種類のTNF受容体スーパーファミリー(TNFRSF)に分類されており,相互作用するリガンドと受容体の組み合わせがあることも解明されている(図13).また,特に,アポトーシスに関連する受容体として,Death domain(DD)を有するTNFRSF1A(TNFR1)やTNFRSF6(FAS)などが同定されている.TNFSFやTNFRSFなどが胸腺細胞のどのサブポピュレーションで発現しており,どのような発現パターンを示しているかは不明であるが,実際にすべてのAire+細胞は,DDを有する受容体に対するリガンド群(TnfLsf-1, -2, -6, -8, -10, -15, Eda1)を発現していた(図14).
このように免疫寛容の成立には,自己抗原を認識するチェック機構“ネガティブセレクション”が正常に働くことが必須であり,この機構が崩壊することによって自己免疫疾患が発症する.
10. Aire+細胞はself-APCの特徴を持つ
Aire+細胞の特徴をまとめると図15のようになる.
通常,ネガティブセレクションをつかさどる胸腺髄質上皮細胞では,プロセスされた自己抗原(ペプチド)がMHCを介して細胞表面に提示される.この自己抗原に反応する“自己に不利な”TCRを発現している胸腺細胞ではアポトーシスが誘発され,除去(負の選択)される.
健常な胸腺の髄質上皮細胞内では,AIREが転写因子として,各種の臓器に特異的な遺伝子群(自己抗原)の発現を亢進し,結果的に,標的臓器に対する免疫寛容が成立している.
一方,自己免疫疾患APECEDにおいては,突然変異によりAIRE遺伝子の機能が失われているので,AIRE依存的な遺伝子群の発現が低下あるいは欠損する.そのため,それら自己抗原タンパク質が翻訳・提示されないことにより,標的自己抗原に反応する胸腺細胞(T細胞)が除去されずに,末梢組織に搬出され,標的細胞(臓器)が攻撃されて,自己免疫疾患が発症する.
T細胞で発現しているTCRは,さまざまな抗原分子に対応できるように多様性に富んでいるが,それらはTCR遺伝子の可変部領域に相当する部分が“ランダム”に再構成されることにより行われている.このようにTCRは,ランダムに再構成されるため,“自己に不利な”自己抗原に反応するものも作られている.
“胸腺クロストーク”とは,胸腺細胞と胸腺ストロマ細胞の間で起こる,双方向のシグナル交換システムのことをいう.つまり胸腺細胞の成熟・分化にはストロマ細胞からのシグナルが必要であり,また,ストロマ細胞の網目状構造の形成には胸腺細胞の助けが必要である.
胸腺細胞からT細胞への成熟や胸腺皮質上皮細胞(cTEC)や髄質上皮細胞(mTEC)の分化において,経時的空間的な複雑かつ精妙な相互作用が営まれている.しかし,現在のところ,胸腺内でのTNFSFとTNFRSFの相互作用はほとんどわかっていない.最近では,CD4+胸腺細胞で発現しているCD40L(TNFSF5)やRANKL(TNFSF11)などのTNFSFメンバーが,実際にAIRE発現mTECの生存運命を決定していることなど,胸腺内におけるTNFSF/TNFRSF機構が証明されつつある19–21).また,いくつかのTNFアンタゴニストが,RAや強直性脊椎炎などの自己免疫疾患の治療薬として用いられているので,TNFSF/TNFRSFが自己免疫疾患の新たな治療薬開発の標的分子になることは十分考えられる.
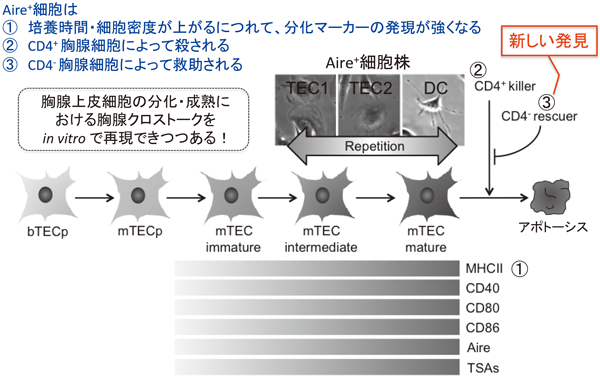
近年,Aireを発現する胸腺の髄質上皮細胞は,その前駆細胞から,immature stage→intermediate stage→mature stageへと分化することがわかってきた.Aireや末梢組織に特異的な遺伝子群,CD80,CD86,CD40,MHC-II等の細胞表面マーカーの発現が増大し,最期はアポトーシスに陥るという胸腺髄質上皮細胞の分化モデル“Terminal Differentiation Model”が,提唱されている22,23).
12. Aire+細胞株を用いて胸腺クロストークをin vitroで再現できるか?
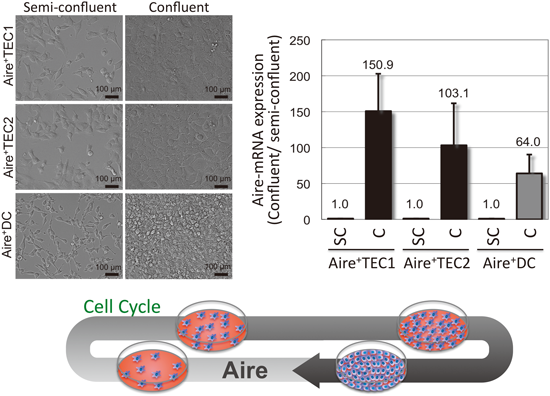
我々は,このモデルをin vitroで検証するために,Aire+細胞の培養に際して,分化マーカーの発現が培養時間や培養環境の違いにより変動するかどうか調べた.このために,培養時間が異なる細胞密度が低いsemi-confluent(SC)状態と高密度のconfluent(C)状態のAire+細胞培養プレートを用意して,Aireや末梢組織特異的自己抗原遺伝子,細胞表面マーカーなどの発現を調べた.その結果,SC→Cと培養時間や細胞密度が高くなるにつれて,Aire遺伝子の発現は約100(64~160)倍も上昇した24)(図16).
また,5種の末梢組織特異的自己抗原遺伝子の発現も再現性よく有意に(1.3~766.7倍)上昇した.さらに,4種の細胞表面マーカーも発現上昇が確認された.つまり,Aire+細胞は,培養時間に伴った細胞密度によって,immature stage→intermediate stage→mature stageへの分化を繰り返すことが示唆された.
13. 胸腺クロストークにおけるAire+細胞と胸腺細胞の挙動
AIREを発現する胸腺の髄質上皮細胞は,ネガティブセレクションにより自己反応性の胸腺細胞を除去している“教育者”であるが,一方,Aire+細胞は特殊な(CD4+3−)胸腺細胞にその運命を左右されている.そこで,我々は,上述のin vitro胸腺クロストーク系における,Aire+細胞の挙動を調べた.
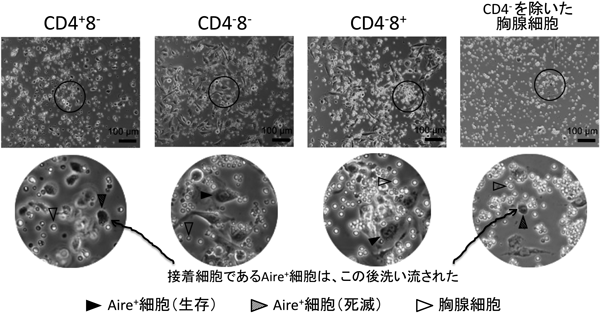
まず,新鮮な胸腺を破砕して得たバルクの胸腺細胞から,マグネットビーズで標識した特異抗体を用いて「CD4+8−」,「CD4−8−」,「CD4−8+」,「CD4−を除いた胸腺細胞」の4群を分離し,それらをAire+細胞と共培養してみた.その結果,「CD4−8−」,「CD4−8+」胸腺細胞との共培養では,Aire+細胞はそれら胸腺細胞にアポトーシスを起こさせたが,自身は死ななかった.これに対し,「CD4+8−」と「CD4−を除いた胸腺細胞(つまりはCD4+)」との共培養においては,逆にAire+細胞が殺された.この現象は,三つのAire+細胞株で同様に観察された24)(図17).
そもそもAire+細胞は,CD4+もCD4−も共存するバルクの胸腺細胞とともに培養しても殺されないが,CD4−を除いたCD4+胸腺細胞群と共培養すると殺された.用いたCD4+胸腺細胞画分には,Aireを発現するmTECの命運(死ぬか生きるか)を決定するCD40L/RANKL陽性CD4+3−胸腺細胞も含まれており,CD4−が「CD4+胸腺細胞によるAire+細胞の死」を回避させていると考えられた.すなわち,Aireを発現する胸腺髄質上皮細胞に対して「CD4+ killer thymocyte」と「CD4− rescuer thymocyte」の2種が存在すると考えた.
このように,Aire+細胞株を用いて胸腺上皮細胞の分化・成熟における胸腺細胞とのクロストークをin vitroで再現できつつある.
一般に,ある細胞タイプにおける特定タンパク質の機能を解析する場合,それが内在的に発現される細胞株(または,濃縮純化した細胞群)を用いることが理想である.しかし,AIREを発現する細胞は,胸腺内の髄質上皮細胞内でもきわめてわずかしか存在せず,細胞表面マーカーの発現パターンが複雑であることから,AIREを発現しているmTECのみを純化させ,それらを大量に準備することができなかったし,AIREを発現している胸腺髄質上皮細胞株も存在していなかったため,in vitroによるAIRE遺伝子・タンパク質の機能解析は,永らく困難なままであった.
我々は樹立したAire+細胞を用いて,胸腺細胞へのアポトーシスの誘発や末梢組織特異的自己抗原遺伝子の異所性発現,免疫プロテアソームの発現などをin vitroで再現・確認できた.さらに,Aire+細胞のさまざまな特徴が,in vivoで観察されているAire発現胸腺髄質上皮細胞の挙動と非常に似ていることも確認した24)(図18).
これらより,胸腺の髄質上皮細胞において,AIREタンパク質が多くの自己抗原となる遺伝子を異所性に発現する機構などをゲノムレベルでさらに解明(図19)することは,APECEDなどの自己免疫疾患の発症機構の解明のみならず免疫寛容の成立機構の解明にも大きく貢献すると考えている.
Aire+細胞は,今後,胸腺の髄質上皮細胞における真のAIREパートナー(協同して転写を制御する因子)の同定やタンパク質レベルでの標的自己抗原の網羅的解析,胸腺クロストークに関与しうるTNFSF/TNFRSFの分子間相互反応機構の解明,胸腺髄質上皮細胞独特のゲノム修飾様態によるAire依存性自己抗原遺伝子群の同定,また異所性発現制御機構の解明等に貢献できるものと考えており,これらを統合した自己免疫疾患の画期的な治療法の開発にも役立つと期待している.
引用文献References
1) Nagamine, K., Peterson, P., Scott, H.S., Kudoh, J., Minoshima, S., Heino, M., Krohn, K.J., Lalioti, M.D., Mullis, P.E., Antonarakis, S.E., Kawasaki, K., Asakawa, S., Ito, F., & Shimizu, N. (1997) Nat. Genet., 17, 393–398.
2) Ono, M., Yaguchi, H., Ohkura, N., Kitabayashi, I., Nagamura, Y., Nomura, T., Miyachi, Y., Tsukada, T., & Sakaguchi, S. (2007) Nature, 446, 685–689.
3) Kogawa, K., Kudoh, J., Nagafuchi, S., Ohga, S., Katsuta, H., Ishibashi, H., Harada, M., Hara, T., & Shimizu, N. (2002) Clin. Immunol., 103, 277–283.
4) Aaltonen, J., Björses, P., Sandkuijl, L., Perheentupa, J., & Peltonen, L. (1994) Nat. Genet., 8, 83–87.
5) Björses, P., Aaltonen, J., Vikman, A., Perheentupa, J., Ben-Zion, G., Chiumello, G., Dahl, N., Heideman, P., Hoorweg-Nijman, J.J., Mathivon, L., Mullis, P.E., Pohl, M., Ritzen, M., Romeo, G., Shapiro, M.S., Smith, C.S., Solyom, J., Zlotogora, J., & Peltonen, L. (1996) Am. J. Hum. Genet., 59, 879–886.
6) Halonen, M., Eskelin, P., Myhre, A.G., Perheentupa, J., Husebye, E.S., Kämpe, O., Rorsman, F., Peltonen, L., Ulmanen, I., & Partanen, J. (2002) J. Clin. Endocrinol. Metab., 87, 2568–2574.
7) Pitkänen, J., Doucas, V., Sternsdorf, T., Nakajima, T., Aratani, S., Jensen, K., Will, H., Vähämurto, P., Ollila, J., Vihinen, M., Scott, H.S., Antonarakis, S.E., Kudoh, J., Shimizu, N., Krohn, K., & Peterson, P. (2000) J. Biol. Chem., 275, 16802–16809.
8) Uchida, D., Hatakeyama, S., Matsushima, A., Han, H., Ishido, S., Hotta, H., Kudoh, J., Shimizu, N., Doucas, V., Nakayama, K.I., Kuroda, N., & Matsumoto, M. (2004) J. Biol. Chem., 279, 33984–33991.
9) Muratani, M. & Tansey, W.P. (2003) Nat. Rev. Mol. Cell Biol., 4, 192–201.
10) Musco, G. & Peterson, P. (2008) Epigenetics, 3, 310–314.
11) Koh, A.S., Kuo, A.J., Park, S.Y., Cheung, P., Abramson, J., Bua, D., Carney, D., Shoelson, S.E., Gozani, O., Kingston, R.E., Benoist, C., & Mathis, D. (2008) Proc. Natl. Acad. Sci. USA, 105, 15878–15883.
12) Org, T., Rebane, A., Kisand, K., Laan, M., Haljasorg, U., Andreson, R., & Peterson, P. (2009) Hum. Mol. Genet., 18, 4699–4710.
13) Anderson, M.S., Venanzi, E.S., Klein, L., Chen, Z., Berzins, S.P., Turley, S.J., von Boehmer, H., Bronson, R., Dierich, A., Benoist, C., & Mathis, D. (2002) Science, 298, 1395–1401.
14) Liston, A., Lesage, S., Wilson, J., Peltonen, L., & Goodnow, C.C. (2003) Nat. Immunol., 4, 350–354.
15) Jiang, W., Anderson, M.S., Bronson, R., Mathis, D., & Benoist, C. (2005) J. Exp. Med., 202, 805–815.
16) Kuroda, N., Mitani, T., Takeda, N., Ishimaru, N., Arakaki, R., Hayashi, Y., Bando, Y., Izumi, K., Takahashi, T., Nomura, T., Sakaguchi, S., Ueno, T., Takahama, Y., Uchida, D., Sun, S., Kajiura, F., Mouri, Y., Han, H., Matsushima, A., Yamada, G., & Matsumoto, M. (2005) J. Immunol., 174, 1862–1870.
17) Abramson, J., Giraud, M., Benoist, C., & Mathis, D. (2010) Cell, 140, 123–135.
18) Yamaguchi, Y., Takayanagi, A., Chen, J., Sakai, K., Kudoh, J., & Shimizu, N. (2011) Exp. Cell Res., 317, 2019–2030.
19) Irla, M., Hugues, S., Gill, J., Nitta, T., Hikosaka, Y., Williams, I.R., Hubert, F.X., Scott, H.S., Takahama, Y., Holländer, G.A., & Reith, W. (2008) Immunity, 29, 451–463.
20) Hikosaka, Y., Nitta, T., Ohigashi, I., Yano, K., Ishimaru, N., Hayashi, Y., Matsumoto, M., Matsuo, K., Penninger, J.M., Takayanagi, H., Yokota, Y., Yamada, H., Yoshikai, Y., Inoue, J., Akiyama, T., & Takahama, Y. (2008) Immunity, 29, 438–450.
21) Akiyama, T., Shimo, Y., Yanai, H., Qin, J., Ohshima, D., Maruyama, Y., Asaumi, Y., Kitazawa, J., Takayanagi, H., Penninger, J.M., Matsumoto, M., Nitta, T., Takahama, Y., & Inoue, J. (2008) Immunity, 29, 423–437.
22) Villaseñor, J., Besse, W., Benoist, C., & Mathis, D. (2008) Proc. Natl. Acad. Sci. USA, 105, 15854–15859.
23) Irla, M., Hollander, G., & Reith, W. (2010) Trends Immunol., 31, 71–79.
24) Yamaguchi, Y., Kudoh, J., Yoshida, T., & Shimizu, N. (2014) Biol. Open, 3, 1071–1082.
著者紹介Author Profile
 山口 良考(やまぐち よしたか)
山口 良考(やまぐち よしたか)慶應義塾大学先導研GSPセンター特任講師.博士(医学).
略歴1970年愛知県生まれ.高校卒業後寿司職人を経て97年杏林大学保健学部臨床検査技術学科卒業.99年同大学院保健学研究科修士課程修了.2003年慶應義塾大学大学院医学研究科満期退学.03~10年慶應義塾大学医学部分子生物研究室助教.11~15年同大学先導研GSPセンター特任助教.13年同大学院医学研究科博士号(医学)取得.15年より現職.
研究テーマと抱負自己免疫疾患APECEDの原因遺伝子AIREの機能解析を志す.独自に樹立したマウスAire+細胞株を用いて,Autoimmunity治療法の確立も目指す.
趣味もっぱら研究と家族.