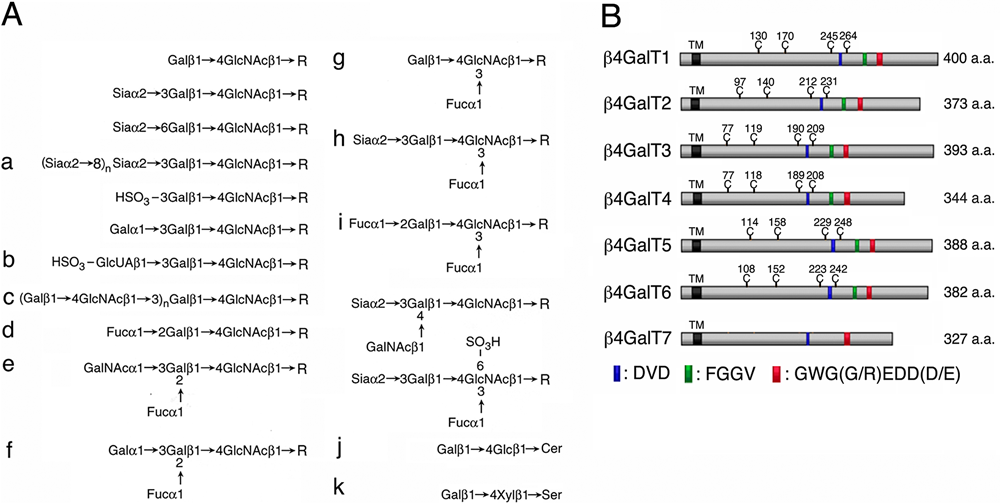
1970年代にL. Warrenら,さらにP. Robbinsらにより,細胞ががん化するとタンパク質に結合したN型糖鎖の分子量が増大することが見いだされ,この現象は1980年代に木幡陽らにより糖鎖の枝分かれ構造の亢進(高分岐化)によることが明らかにされた1,2).特にN型糖鎖では,Man3GlcNAc2からなる母核構造のManα1→6Man分岐の非還元末端マンノース(Man)にβ-1,6結合でN-アセチルグルコサミン(GlcNAc)が結合したGlcNAcβ1→6Man分岐が亢進する.いずれの分岐側鎖のGlcNAc残基もさらに複数のβ4-ガラクトース転移酵素(β4GalT)によりガラクトース(Gal)が付加され,Galβ1→4GlcNAc側鎖が形成される.O型糖鎖に関しても,細胞ががん化すると分岐構造を持つコア2糖鎖Galβ1→4GlcNAcβ1→6(Galβ1→3)GalNAcが増加する2).興味深いことに,発生やがん化に関連した抗原の多くはGalβ1→4GlcNAc側鎖を土台として複数の糖が付加されて形成され(図1A),細胞間の接着や転移などに関与することが知られている3).一方,糖脂質ではシアル酸を含むガングリオシドGM3,GD3が一般的に悪性度の高い脳腫瘍やメラノーマで高発現する.GM3やGD3は,ラクトシルセラミドGalβ1→4Glcβ1→Cer(Lac-Cer)を土台として合成される.この他ラクト,ネオラクト,グロボ,イソグロボ系列の糖脂質もLac-Cerを土台として合成されるので,Lac-Cerの発現調節は糖脂質の機能発現にきわめて重要である.このLac-Cerの生合成には,β4GalT5とβ4GalT6が関与している4).
これらの糖タンパク質や糖脂質は細胞表面に多く存在しているので,タンパク質や脂質に結合した糖鎖でのガラクトースの発現動態は細胞の性質を左右しうることが想像される.本稿ではまず,β4GalT遺伝子の発現を変化させることでがん細胞表面のガラクトースが細胞の性質を制御していることを示し,さらにβ4GalT遺伝子の遺伝子治療への応用の可能性を紹介したい.
2. β4-ガラクトース転移酵素(β4GalT)ファミリーと基質特異性
β4GalTには構造の似た七つのアイソザイムが存在し(β4GalT1~7),これら酵素に見いだされるいくつかの相同性領域はβ4GalTファミリーに共通の基質や糖供与体を認識する部位と考えられている(図1B)4).これら7種の酵素は基質特異性(分岐側鎖など),発生段階での発現パターン,発現組織などに違いがあることが知られている.特に基質特異性に関しては完全には解析されておらず,さらなる解析が必要である.これまでに得られている報告から,β4GalT1とβ4GalT2はN型糖鎖の分岐側鎖に幅広くガラクトースを転移し,特にポリ-N-アセチルラクトサミン(図1A-c)の合成に高い活性を持つ.さらに,β4GalT2はα-ジストログリカンに結合したGlcNAcβ1→2Man鎖やNotch受容体に結合したGlcNAcβ1→3Fuc鎖に効率的にガラクトースを転移する.β4GalT3は糖脂質上のポリ-N-アセチルラクトサミノグリカンの合成において,最初のN-アセチルラクトサミン構造の合成に関与し,β4GalT4はネオラクト系列の糖脂質合成に関与している.最近の研究ではβ4GalT3はN型糖鎖のもつポリ-N-アセチルラクトサミンの合成に,またβ4GalT4はN-アセチルグルコサミン-6-硫酸のガラクトシル化を行うことができ,ケラタン硫酸の生合成に関与している可能性が報告されている.さらに,β4GalT4はセレクチンのリガンドとなるSiaα2→3Galβ1→4(Fucα1→3)(SO3→6)GlcNAc抗原を合成する酵素の可能性も考えられている.我々はヒト乳がん細胞からβ4GalT5遺伝子をクローニングし(当時は糖鎖のガラクトシル化は古典的なβ4GalTIのみしか存在しないと考えられていたのでβ4GalTIIと命名した)5),in vitroで酵素活性を測定するとN型糖鎖やO型糖鎖のN-アセチルグルコサミンのみならず,グルコシルセラミド(Glc-Cer)へも効率よくガラクトースを転移することを見いだした.この酵素遺伝子をがん細胞から単離したこと,分岐構造を持つ基質糖鎖へガラクトースを転移することから,我々はβ4GalT5が糖タンパク質糖鎖のガラクトシル化に関与するがん関連遺伝子であると考えていた.しかしながらGlc-Cerへガラクトースを転移しLac-Cerを合成する酵素(β4GalT6)が精製されその遺伝子もクローニングされると,両タンパク質の間には70%の相同性が存在すること3),さらにβ4GalT5はβ4GalT6より効率よくLac-Cerを合成することから4,6),実際β4GalT5は細胞内でどの分子を基質としているのかが疑問となった.この問題を解決するにはβ4GalT5遺伝子を破壊したマウスの細胞で,糖タンパク質と糖脂質の分析を行うことが必要と考え,β4GalT5−/−マウス胎仔から繊維芽細胞(MEF)を調製し解析を行った.その結果,糖タンパク質糖鎖のガラクトシル化には変化はみられなかったが,Lac-Cerとそれから派生するGM3の発現量がβ4GalT5+/+,β4GalT5+/−,β4GalT5−/−マウス由来のMEF細胞の順に比例して減少し,β4GalT5はLac-Cer合成酵素であることが明らかとなった7).ホモ個体のMEF細胞に見いだされたわずかなLac-Cerは,β4GalT6によるものと思われる.この時点で哺乳動物細胞には二つのLac-Cer合成酵素が存在することが判明したが,その存在意義に関しては現在でも不明なままである.in vitroのアッセイ系では酵素は加えた基質に容易にアクセスできるが,in vivoでは酵素の局在と,その近傍に実際基質が存在するかが合成される分子の重要な決定要因となる.したがって,酵素の存在意義を考えるためには,in vitroのアッセイ系だけでなくその酵素遺伝子不活性化細胞を用いた細胞レベルでの解析が必須である.ちなみにβ4GalT7はGalTIとも呼ばれ,グリコサミノグリカン鎖がコアタンパク質と結合する架橋領域Galβ1→3Galβ1→4Xylの最初のガラクトースを転移する酵素であり,この酵素遺伝子の変異は早期老化症の一つであるEhlers-Danlos症候群を引き起こすことが知られている(各酵素の基質特異性に関する詳細は総説参照4,6)).
細胞ががん化するとGlcNAcβ1→6Man分岐を合成するN-アセチルグルコサミン転移酵素V(GlcNAcTV)の活性や遺伝子発現が増加することが知られていたが,細胞のがん化に伴うβ4GalTの変動についての網羅的な解析は行われていなかった.そこで我々はNIH3T3細胞とそれをポリオーマウイルスのmiddle T抗原遺伝子で形質転換させた細胞をがん化細胞のモデルとしてβ4GalT1~6の遺伝子発現を解析した.その結果,middle T抗原遺伝子の導入によりβ4GalT2遺伝子の発現が低下し,β4GalT5遺伝子の発現が増加することを見いだした8).さらにヒト由来の複数のがん細胞株でGlcNAcTVと二つのβ4GalT遺伝子の発現を比較すると,β4GalT2遺伝子の発現はGlcNAcTVの発現と反比例の関係に,β4GalT5遺伝子の発現は正比例の関係にあることが判明した9).
ヒト白血病細胞ではがんの悪性度の指標の一つである薬剤耐性の獲得に,β4GalT1とβ4GalT5の遺伝子発現の増大が関与している.これらの遺伝子発現が高くなるとヘッジホッグシグナルが活性化され,膜輸送タンパク質であるP糖タンパク質や薬剤耐性関連タンパク質の発現が増大する.実際,HL60細胞でβ4GalT1あるいはβ4GalT5の遺伝子発現を人工的に高めると細胞は薬剤耐性を獲得すること,逆にβ4GalT1あるいはβ4GalT5の遺伝子発現を抑制すると薬剤耐性細胞が薬剤への感受性を取り戻すことが報告されている10).糖鎖がどのように機能しているのかはわからないがきわめて興味深い知見である.HeLa細胞でβ4GalT2の遺伝子発現を高めるとp53を介したアポトーシスが誘導されること,さらにβ4GalT2遺伝子の転写因子がp53であることから11),がん細胞ではp53を不活性化し,その結果β4GalT2遺伝子の発現が抑制され細胞のアポトーシスを防いでいると考えられる.またβ4GalT3に関して,遺伝子の高発現は脳腫瘍で細胞の移動や浸潤を促進し,一方ヒト直腸がんでは細胞の転移を抑制することが報告されている12,13).このような相違はがん細胞で固有のガラクトシル化が特定の糖タンパク質で起き,さらに固有の糖脂質が発現してくるためであると考えられ,これらがどのような現象を支配しているのか解析が必要である.
4. がん細胞におけるβ4GalT遺伝子発現の改変とその腫瘍形成能の変化
1)β4GalT2遺伝子の発現増強
細胞ががん化するとβ4GalT2遺伝子の発現が低下する.そこで悪性度の高いB16-F10マウスメラノーマ細胞からβ4GalT2高発現細胞株を数株樹立した.これらの細胞はいずれも複数の細胞膜タンパク質で糖鎖のガラクトシル化が亢進しており,さらに細胞密度が高くなると全体に細胞と細胞の境界が明確になり,細胞間接着が回復してきたと思われた.また,β4GalT2遺伝子の発現が対照の細胞株と比較して2.5倍高いD4細胞株と対照の細胞株をC57BL/6マウス皮下に移植すると,D4細胞でその腫瘍形成が著しく抑制された(表1A).形成された腫瘍組織を免疫組織化学的に解析すると,D4細胞由来の腫瘍ではTUNEL陽性細胞が出現しアポトーシスが誘導され,さらにCD31陽性の血管内皮細胞が減少し血管新生が抑制されていることが見いだされた14).これらの現象にはガレクチンが関与することが知られており,B16-F10細胞でβ4GalT2遺伝子の発現が増大すると細胞膜タンパク質に結合した糖鎖のガラクトシル化が亢進し,特定のガレクチンが結合あるいは結合できなくなり,上記の現象が誘導されたものと考えられる.
表1 β4GalT2センスcDNA(A)またはβ4GalT5アンチセンスcDNA(B)を導入したB16-F10マウスメラノーマ細胞株の造腫瘍能 | 細胞株 | 腫瘍形成動物の匹数 | 腫瘍径(mm)(n) | p-値 |
|---|
| (A) | B16-mock (C2) | 10/10 | 13.1±0.8 (10)b | |
| B16-β4GalT2 (D4) | 8/10 | 5.1±1.1 (8) | <0.01a |
| (B) | B16-mock (C1) | 10/10 | 13.3±0.5 (10)d | |
| B16-β4GalT5 (E4) | 10/10 | 10.4±0.6 (10) | <0.05c |
| B16-β4GalT5 (E5) | 9/10 | 5.9±0.7 (9) | <0.01c |
| a C2細胞とD4細胞が造る腫瘍の平均サイズの比較における有意差.c C1細胞とE4細胞およびC1細胞とE5細胞が造る腫瘍の平均サイズの比較における有意差.b, d 数値は平均長±標準誤差(匹数).(A)は文献14からNature Publishing Groupの転載許可を,また(B)は文献15からOxford University Pressの転載許可を得て一部変更し掲載. |
2)β4GalT5遺伝子の発現抑制
細胞ががん化するとβ4GalT5遺伝子の発現が増大する.そこで上記と同様にB16-F10細胞へβ4GalT5アンチセンスcDNAを導入し,β4GalT5遺伝子の発現を抑制した細胞株を数個得た.これらの細胞ではLac-Cerの発現量が減少するとともに,Lac-Cerから派生するGM3の発現量も減少していた.さらにこれらの細胞は培養系での増殖速度も減少し,細胞密度が高くなると細胞間の接着が部分的にみられた.細胞株の中で対照と比較してβ4GalT5遺伝子の発現が23%,65%抑制されたE4, E5細胞株と対照の細胞株をそれぞれC57BL/6マウス皮下に移植すると,E4,E5細胞株での腫瘍の形成が対照株と比較して有意に抑制され,その抑制度はβ4GalT5遺伝子の発現抑制度と相関する傾向がみられた(表1B).この腫瘍を上記と同様に免疫組織化学的に解析すると,アポトーシスの誘導や血管新生の阻害に若干の変化はみられるものの,むしろがん細胞で増殖を促進するMAPK経路の情報伝達分子のリン酸化が抑制されていることが見いだされた15).Lac-CerはMAPK経路を活性化することが知られているので,β4GalT5遺伝子の発現抑制がLac-Cer合成を抑制した結果と思われる.
上記の実験から,がん細胞の腫瘍形成能とβ4GalT5遺伝子の発現レベルとの間に相関があると考えられた.しかし,遺伝子を導入した細胞では導入遺伝子の脱落などが起こり,安定した状態で維持することが難しいので,元々のβ4GalT5遺伝子量の異なるβ4GalT5−/−,β4GalT5+/−,β4GalT5+/+マウス由来のMEF細胞にポリオーマウイルスのmiddle T抗原遺伝子を導入して細胞をがん化させ,その性質を解析した.まずこれらの細胞の形質転換率を軟寒天培地で増殖するコロニーの大きさ(体積)と数で解析すると,その大きさと数はβ4GalT5−/−<β4GalT5+/−<β4GalT5+/+マウス由来のMEF細胞の順で高かった.次にこれらの細胞をヌードマウス皮下に移植し腫瘍形成能を解析すると,上記の順に大きかった.以上の結果から,がん細胞の腫瘍形成能はβ4GalT5の遺伝子量(コピー数)に比例すると結論づけられた15).
しかしながら,この現象ではβ4GalT5遺伝子の最終産物であるLac-Cer自身が重要であるのか,それともLac-Cerから派生するGM3等の多様な糖脂質が重要であるのかは不明である.ヒト好中球ではLac-Cerは細胞膜の脂質ラフトに局在し細胞膜の機能を制御している知見があるので16),一般のがん細胞でもLac-Cerが脂質ラフトに存在し,そこに局在する細胞接着分子や増殖因子受容体の機能を制御している可能性が考えられる.実際,対照細胞に比べフィブロネクチンへの接着性が著しく低いβ4GalT5−/−マウス由来のMEF細胞をLac-Cerを含む培地で培養し細胞膜に取り込ませるとフィブロネクチンへの接着性が回復することから,Lac-Cer自身が細胞膜でインテグリン分子の機能を制御していることが考えられる(論文投稿準備中).細胞膜でLac-Cerの発現量が増えるとそれがどのような働きで細胞の性質を変えうるのか,そのメカニズムの解明が今後重要であり,それは細胞の異常な増殖のメカニズムを解き明かす新たな出発点となるかもしれない.
Lac-Cerを合成する活性を有する酵素としてβ4GalT6も存在するが,その発現レベルはβ4GalT5と比較し低く,かつ細胞のがん化で遺伝子発現の変動はみられない.β4GalT5のノックアウトマウスは胎生致死となるが,β4GalT6のノックアウトマウスには顕著な異常はみられず生存できる.これら二つの酵素で合成されるLac-Cerに脂肪酸の構造の違いを含めた相違があるのか,これもきわめて興味深い問題である.最近,多発性硬化症(中枢性脱髄疾患)モデルマウスの炎症部位でβ4GalT6によるLac-Cerの生合成が亢進し,これが炎症を促進することが報告されている17).このLac-Cerはβ4GalT5が生合成するものと質的に異なるのか,あるいはこの炎症部位でβ4GalT6遺伝子の発現が特異的に活性化されているのか,その転写制御のメカニズムなどを解明する必要がある.
3)二つのβ4GalT遺伝子の発現の改変
これまで述べてきたようにB16-F10細胞でβ4GalT2遺伝子の発現を増大あるいはβ4GalT5遺伝子の発現を低下させると,いずれも腫瘍形成が抑制され,これらは異なるメカニズムで腫瘍の増殖を抑制していることが判明した.またこれらの遺伝子発現の変化は細胞のがん化に伴い観察されるので,究極的にはがん細胞で二つの遺伝子の発現を同時に制御すれば,より腫瘍形成を抑制できると考えられる.実際,B16-F10細胞で二つの遺伝子を操作すると,単独で遺伝子を操作した細胞よりさらに腫瘍形成が抑制される結果が得られている(論文投稿準備中).
5. β4GalT遺伝子によるヒト腫瘍の増殖抑制の試み
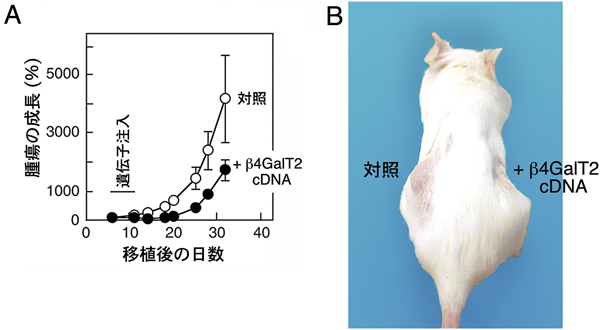
特定の遺伝子を導入しその発現が安定したがん細胞株を動物に移植してその腫瘍形成を抑制できても,発生してきた腫瘍へ遺伝子を均一に効率高く導入することは難しい.何とかしてβ4GalT遺伝子を標的としたヒトのがん治療はできないものであろうか.そこで千葉県がんセンター病理部門の田川雅敏博士と共同研究を行い,SCIDマウス皮下にヒト肝臓がん細胞を移植し5 mm大の腫瘍を造り,この腫瘍へアデノウイルスベクターに組み込んだヒトβ4GalT2 cDNAを注射器で複数箇所へ注入し,腫瘍の増殖を観察した.その結果,腫瘍の増殖は対照(アデノウイルスベクターのみ注入)に比べて有意に抑制された(図2)14).これらの腫瘍組織ではTUNEL陽性細胞の増加やCD31陽性細胞の減少がみられたが,腫瘍全体で統計処理を行うと遺伝子が注入されなかった領域も含まれるため有意差は得られなかった.しかし,このことはもしヒトβ4GalT2 cDNAを腫瘍へ均一に注入できる手法を開発すれば,その増殖を有意に抑制することができることを示している.またβ4GalT5遺伝子に関しても,そのアンチセンスcDNAやsiRNAを腫瘍に注入し効率的にβ4GalT5の発現レベルを低下させれば,同様に腫瘍の増殖を抑制できる.細胞のがん化ではタンパク質と脂質に結合した糖鎖に同時に変化が起こるので,β4GalT2とβ4GalT5の二つの遺伝子の発現を制御することが肝要である.
1990年に米国でアデノシンデアミナーゼ(ADA)欠損による重症複合免疫不全症(SCID)の患者に初めて遺伝子治療が行われた.これは患者の血液から造血幹細胞を取り出し遺伝子を導入し,体内へ戻すことにより行われた.1995年に同じ疾患の患者で国内でも遺伝子治療が行われた.しかしながら,その後大量のベクター(遺伝子の運び屋)を用いたことによる死亡例や,2002年にフランスで実施されたX染色体連鎖重度免疫不全症患者への遺伝子治療で白血病などの副作用が引き起こされたことから,遺伝子治療は据え置かれてきた.その後新たなベクターの開発が進み,遺伝子治療の有効性を示す研究も増え,欧州では2012年に初めて遺伝子治療薬としてGlyberaが承認され,リポタンパクリパーゼ(LPL)欠損症の患者に投与されている.本稿で述べたように細胞のがん化で生じる糖鎖の構造異常を修復すると腫瘍の増殖を多方面から抑制できることから,本糖鎖遺伝子(糖鎖を生合成する酵素をコードする遺伝子)はがんの遺伝子治療薬として大いに期待できる.
引用文献References
1) Kobata, A. (1989) Pigment Cell Res., 2, 304–308.
2) Bhavanandan, V.P. & Furukawa, K. (1995) Biology of the Sialic Acids (Rosenberg, A., ed.), pp. 145–196, Plenum Press, New York.
3) Furukawa, K. & Sato, T. (2000) Biochim. Biophys. Acta, 1473, 54–66.
4) Furukawa, K., Clausen, H., & Sato, T. (2014) Handbook of Glycosyltransferases and Related Genes (2nd Ed.) (Taniguchi, N., Honke, K., Fukuda, M., Narimatsu, H., Yamaguchi, Y., & Angata, T., eds.), pp. 63–72, Springer, Tokyo.
5) Sato, T., Furukawa, K., Bakker, H., Van den Eijnden, D.H., & Van Die, I. (1998) Proc. Natl. Acad. Sci. USA, 95, 472–477.
6) Furukawa, K. (2014) Proc. Jpn. Acad. Sci. Series B, 91, 1–16.
7) Kumagai, T., Sato, T., Natsuka, S., Kobayashi, Y., Zhou, D., Shinkai, T., Hayakawa, S., & Furukawa, K. (2010) Glycoconj. J., 27, 685–695.
8) Shirane, K., Sato, T., Segawa, K., & Furukawa, K. (1999) Biochem. Biophys. Res. Commun., 265, 434–438.
9) Sato, T., Shirane, K., Kido, M., & Furukawa, K. (2000) Biochem. Biophys. Res. Commun., 276, 1019–1023.
10) Zhou, H., Ma, H., Wei, W., Ji, D., Song, X., Sun, J., Zhang, J., & Jia, L. (2013) Cell Death Dis., 4, e654.
11) Zhou, J., Wei, Y., Liu, D., Ge, X., Zhou, F., Yun, X., Jiang, J., & Gu, J. (2008) J. Biochem., 143, 547–554.
12) Chang, H.-H., Chen, C.-H., Chou, C.-H., Liao, Y.-F., Huang, M.-J., Chen, Y.-H., Wang, W.-J., Huang, J., Hung, J.-S., Ho, W.-L., Jeng, Y.-M., Che, M.-I., Lee, H., Lu, M.-Y., Yang, Y.-L., Jou, S.-T., Lin, D.-T., Lin, K.-H., Hsu, W.-M., & Huang, M.-C. (2013) Clin. Cancer Res., 19, 1705–1716.
13) Chen, C.H., Wang, S.H., Liu, C.H., Wu, Y.L., Wang, W.J., Huang, J., Hung, J.S., Lai, I.R., Liang, J.T., & Huang, M.C. (2014) Carcinogenesis, 35, 1258–1266.
14) Tagawa, M., Shirane, K., Sato, T., Furukawa, S., Mizuguchi, H., Kuji, R., Kawamura, K., Takahashi, N., Kato, K., Hayakawa, S., Sawada, S., & Furukawa, K. (2014) Cancer Gene Ther., 21, 219–227.
15) Shirane, K., Kuji, R., Tareyanagi, C., Sato, T., Kobayashi, Y., Furukawa, S., Murata, T., Kubota, S., Ishikawa, Y., Segawa, K., & Furukawa, K. (2014) Glycobiology, 24, 532–541.
16) Iwabuchi, K. & Nagaoka, I. (2002) Blood, 100, 1454–1464.
17) Mayo, L., Trauger, S.A., Blain, M., Nadeau, M., Patel, B., Alvarez, J.I., Mascanfroni, I.D., Yester, A., Kivisakk, P., Kallas, K., Ellezam, B., Bakshi, R., Prat, A., Antel, J.P., Weiner, H.L., & Quintana, F.J. (2014) Nat. Med., 20, 1147–1156.
著者紹介Author Profile
 古川 清(ふるかわ きよし)
古川 清(ふるかわ きよし)長岡技術科学大学大学院生物機能工学専攻教授.理学博士.
略歴1949年新潟県に生る.74年静岡大学理学部卒業.79年東京大学大学院修了.同年ペンシルバニア州立大学,82年ペンシルバニア大学,86年東大医科学研究所,94年都老人総合研究所を経て,2005年から現職.
研究テーマと抱負増殖分化における細胞表面糖鎖の機能解明.自己・非自己の認識機構やcontact inhibitionの分子機構に関与する糖鎖の機能解明を通して,なぜ糖鎖には多様な構造が同時に発現しているのかその意味を探りたい.
ウェブサイトhttp://bio.nagaokaut.ac.jp/~furukawa-l/
趣味音楽鑑賞と演奏,史跡探索.