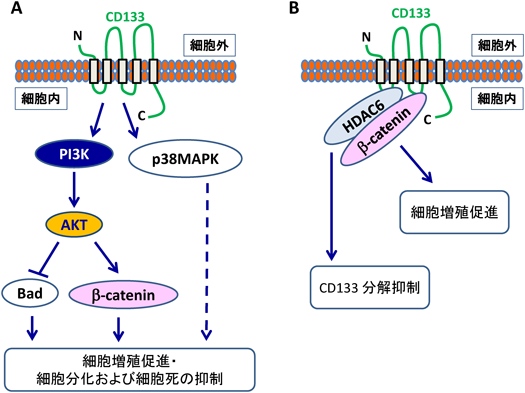
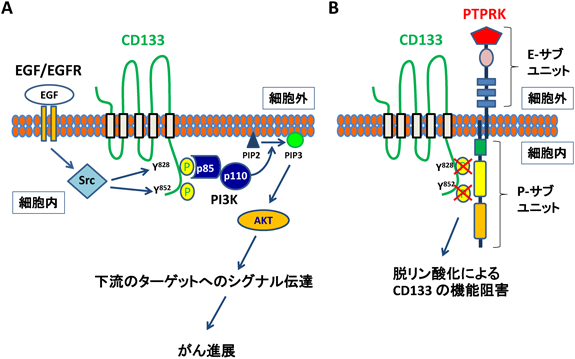
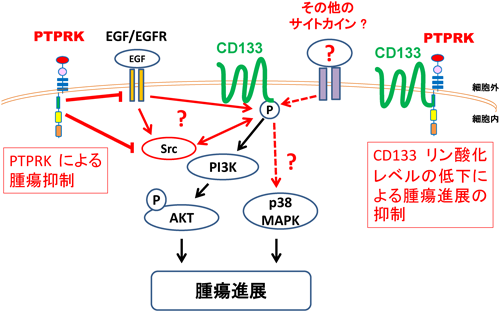
CD133シグナルにおけるチロシンリン酸化の意義The role of tyrosine phosphorylation of cancer stem cell marker CD133 in malignant tumor progression
1 千葉県がんセンター研究所発がん研究グループDNA損傷シグナル研究室Division of DNA Damage Signaling, Carcinogenesis Research Group, Chiba Cancer Center Research Institute (CCCRI) ◇ 〒260-8717 千葉県千葉市中央区仁戸名町666番地の2666-2 Nitona-cho, Chuo-ku, Chiba-shi, Chiba 260-8717 Japan
2 千葉県がんセンター研究所がん治療開発グループがん遺伝創薬研究室Division of Cancer Genetics, Cancer Therapeutic Group, Chiba Cancer Center Research Institute (CCCRI) ◇ 〒260-8717 千葉県千葉市中央区仁戸名町666番地の2666-2 Nitona-cho, Chuo-ku, Chiba-shi, Chiba 260-8717 Japan
発行日:2015年6月25日Published: June 25, 2015