生命とは何か? 答えを一つに限定することはもちろん難しいが,生命の一つの側面として,生命とはゲノム情報から遺伝子調節ネットワークを恒常的・自律的に構築しているシステムである,といっても大きな間違いはないだろう.ここで,セントラルドグマを想起すると,ゲノム・DNA→RNA→タンパク質→ネットワークという流れを考えることになるのであるが,このように矢印(→)を一方向におくことは,ノンコーディングRNA(non-coding RNA)がきわめて盛んに研究されている現状では,もはや不正確であるといわざるをえない.
細胞内には,タンパク質へと翻訳されないRNA(non-coding RNA: ncRNA)が大量に存在する.ncRNA群はその機能,長さなどの観点からさまざまなカテゴリーに分類されるが,これらのncRNAの中で低分子RNAの代表の一つであるmicroRNA(miRNA)は,遺伝子発現の制御において重要な役割を果たしている.miRNAによる遺伝子発現の調節という概念は1993年にすでに報告されているが1,2),今日のmiRNA研究は,1998年のFire,MelloらによるRNA干渉(RNA interference: RNAi)の報告を起点として3),再発見が促され飛躍的に進んだといえる.RNAiは,二本鎖RNAの導入により二本鎖RNAと相補的な塩基配列を持つmRNAが分解される現象であり,20~30塩基からなる内在性の短鎖ncRNAによって制御される遺伝子発現調節機構と合わせてRNAサイレンシングと総称される4,5).RNAiの報告以後,20塩基程度の低分子RNAの探索が行われ6–8),今日miRNAを含む一連の低分子RNAがさまざまな生物種で同定されている.現在,ヒトではすでに1500種以上のmiRNAが報告されている.さらに,miRNAはさまざまな生理現象,および,悪性腫瘍を含むさまざまな疾患の分子病態に関与しており,精力的に研究が行われている9,10).
本稿では,miRNAのユニークな生合成調節機構と遺伝子発現調節機構に関するこれまでの知見を紹介し,未解決の問題とmiRNA生物学から核酸医薬への発展の可能性について議論する.
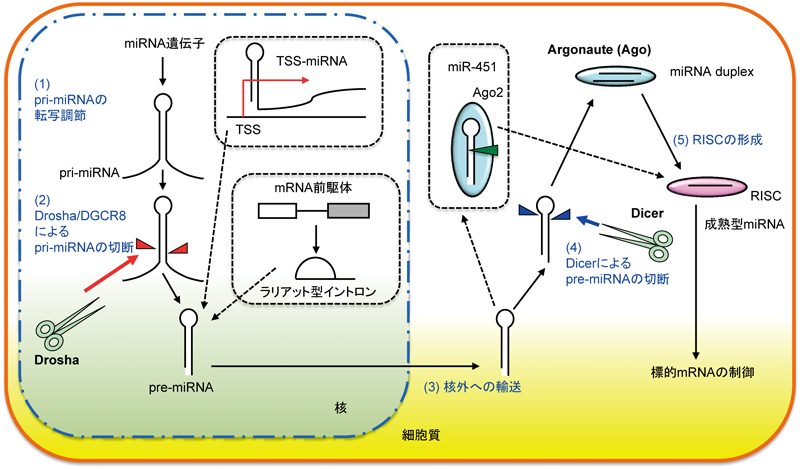
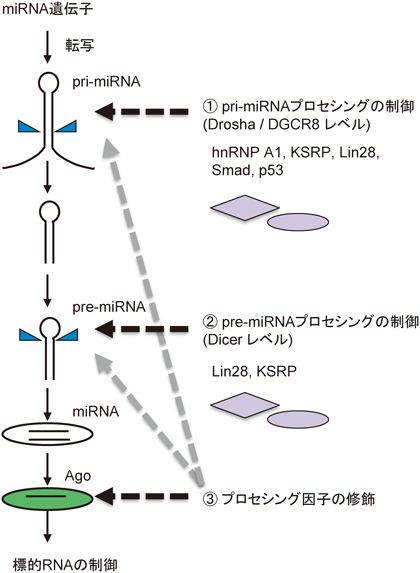
miRNAは21~25塩基程度の短鎖ncRNAであり,内在性のRNAサイレンシング機構を担う代表的な低分子RNA群である4,5,11).標準的なmiRNAの産生過程は,(1)転写によるprimary miRNA(pri-miRNA)の合成,(2)核内でのpri-miRNAの切断,precursor miRNA(pre-miRNA)の産生,(3)pre-miRNAの核外輸送,(4)細胞質でのpre-miRNAの切断,miRNA duplexの産生,(5)RNA誘導型サイレンシング複合体(RNA-induced silencing complex: RISC)の形成,の各ステップからなる4,5,11).これらの過程をmiRNAプロセシングと総称する(図1).
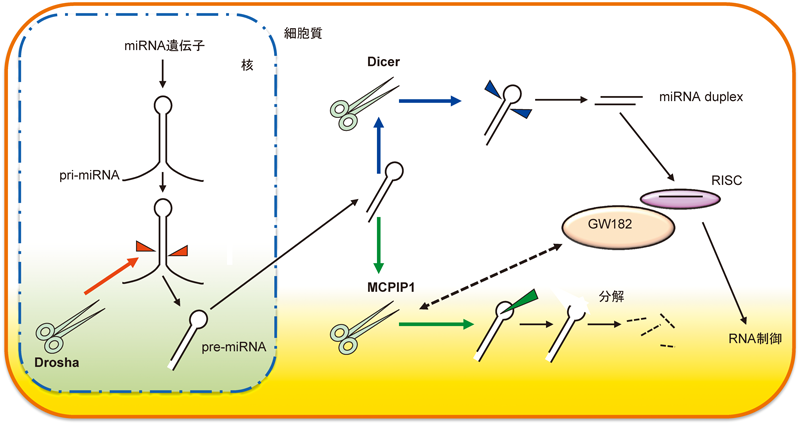
miRNAの前駆体となる最初の転写産物は,タンパク質へ翻訳される遺伝子と同じように,核内において,miRNA遺伝子が主にRNAポリメラーゼⅡにより転写されることによって産生される.これらのヘアピン構造を含む一次転写産物は,primary miRNA(pri-miRNA)と呼ばれる.哺乳動物の約半分のmiRNAは,タンパク質をコードする遺伝子のイントロン領域に存在する.次に,核内でRNaseIIIであるDroshaがpri-miRNAのヘアピン基部を切断することにより,中間産物であるヘアピン構造をした60~70塩基のmiRNA前駆体(precursor miRNA: pre-miRNA)が産生される12,13).DroshaはDGCR8および複数の補助因子と複合体を形成し,ヘアピン構造と一本鎖構造の分岐点から約11塩基離れた部分でヘアピン構造を切断する12,13).Bartelらのグループは,pri-miRNAのヘアピン内部・周囲の特徴的な配列の配置が効率よいプロセシングに重要であることを報告している14).このようにして産生されたpre-miRNAは,主に,exportin-5(XPO5)により核から細胞質に輸送され,別のRNaseIIIであるDicerによって切断され,21~24塩基の二本鎖RNA(miRNA/miRNA* duplex)となる.二本鎖RNAは後述するAgoタンパク質に取り込まれ,片側のRNA鎖(miRNA鎖,ガイド鎖,成熟型miRNA)だけが最終的にAgoタンパク質と安定な複合体を形成し,RNA誘導型サイレンシング複合体(RNA-induced silencing complex: RISC)を形成する15).最終的に,この一本鎖化された成熟型miRNAが遺伝子発現制御のガイド役として働く.
一方で,イントロン自体がpre-miRNAとなるmirtronや,最近報告されたTSS-miRNA(transcriptional start site miRNA)などの産生はDroshaを必要とせず,また,赤血球で発現の高いmiR-451のプロセシングはDicerを必要としないことも報告されており,miRNA生合成経路には多様性が認められる(図1の点線部)11,16,17).
3. miRNAによる遺伝子発現調節機構とその特徴
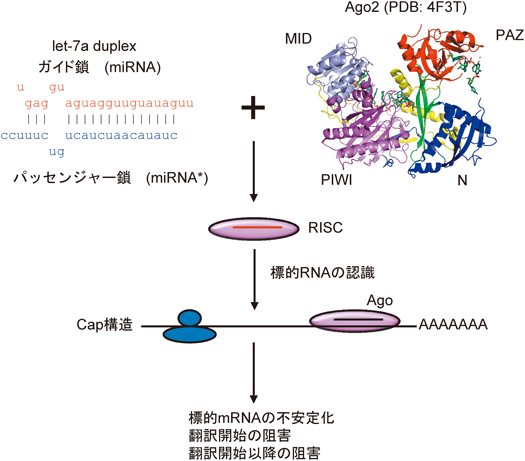
miRNAによる遺伝子調節で中心的な役割を果たすのが,Argonauteファミリーに属するAgoタンパク質である(図2)15,18).miRNAとAgoタンパク質の複合体であるRISCは,RISCに取り込まれたmiRNAと部分的に相補的な配列を有する標的mRNAと結合し,一般的にはその標的mRNAのタンパク質への出力を抑制する.
さまざまな生物種には複数のAgoが存在し[ヒト・マウスで4種(Ago1~4),ショウジョウバエで2種],それぞれのAgoは,標的mRNAの切断活性,miRNA/siRNA(small interfering RNA)の取り込みなどの観点から異なる特徴を有する15,18).哺乳類では,Ago2だけがRNA切断活性(スライサー活性)を有し,siRNAによる標的mRNAの切断を誘導する.Ago1, 3, 4はスライサー活性を有していないが,これは,スライサー活性を担うPIWIドメインの活性中心のアミノ酸が保存されていないことと,N末端の配列の違いによる19–22).ショウジョウバエではmiRNAはAgo1に,siRNAはAgo2に選択的に取り込まれるが,ヒトではmiRNAはAgo1~4すべてに取り込まれており,Ago1, 3, 4もAgo2と同様にmiRNAによる遺伝子抑制機能を仲介している18).
動物細胞におけるmiRNAによる標的mRNAの認識は,主にseed配列と呼ばれる5′末端のわずか7~8塩基と標的mRNAの主に3′非翻訳領域(3′UTR)の相補的塩基配列との間における塩基対形成によって行われる4,23).しかし,一方で,CLIP(cross-linking immunoprecipitation)などの網羅的解析手法の発展とともに,miRNAの結合部位は5′UTRおよびタンパク質コード領域にも存在することが明らかとなっている24–26).miRNAによる標的mRNAの認識は配列特異的であるが,seed配列がきわめて短いこともあり,miRNAとmRNAの対応は「多対多」であり,1種類のmiRNAは複数のmRNAを標的とし,逆に,1種類のmRNAは複数のmiRNAによって制御されることになる4,23,27).miRNAによる遺伝子発現制御のメカニズムとして,(1)標的mRNAの不安定化,(2)翻訳開始の阻害,(3)翻訳開始以降の阻害などのメカニズムがこれまでに提案されている(図2).哺乳類での解析では,これらのメカニズムの相対的な寄与が詳細に検討されており28–30),miRNAによる遺伝子発現抑制は,主にmRNAの不安定化によるものであることが提案されている.
miRNAは多数の標的遺伝子を調節することにより,さまざまな転写因子と同じように,多様な細胞種の正常分化において重要な役割を持つ.細胞機能におけるmiRNAの多様な役割を反映して,miRNAはさまざまな疾患でも重要な役割を果たしている.本稿では,miRNAとがんの関係について注目する.
1)悪性腫瘍におけるmiRNAの役割
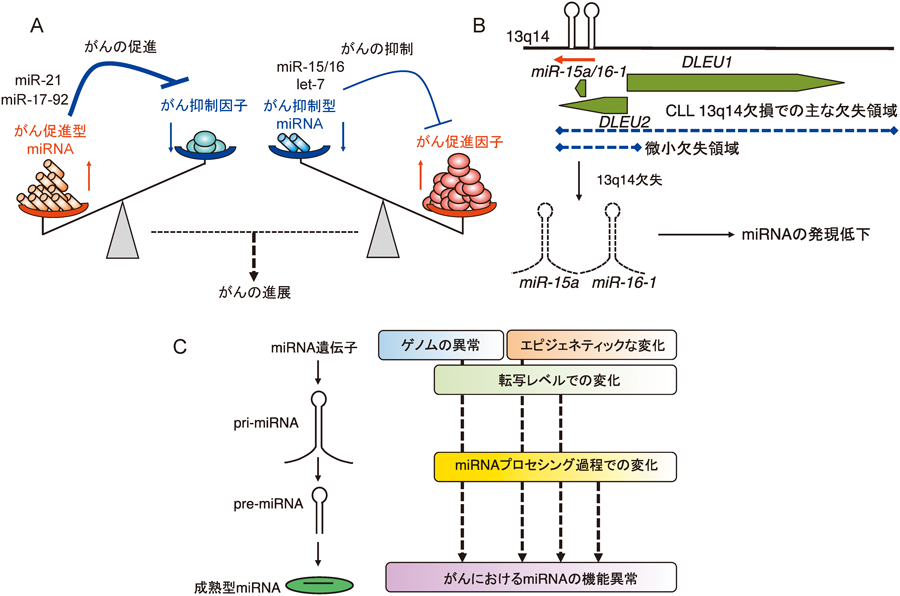
がんとmiRNAの関係は,miRNAによるがん抑制遺伝子・がん遺伝子の制御,がん抑制遺伝子・がん遺伝子によるmiRNAの制御の大きく二つの視点から,検証が行われてきた.慢性リンパ性白血病(chronic lymphocytic leukemia: CLL)で発現低下がみられるmiR-15a/miR-16-1がBcl-2を制御すること,肺がんで発現低下がみられるlet-7がRasがん遺伝子を阻害すること,リンパ腫や肺がんで過剰発現が認められるmiR-17-92クラスターががん遺伝子のように悪性腫瘍を促進するといった重要な報告が2000年代前半になされている9,10,31–34).さまざまながん種で発現の異常を示すmiRNAが報告されており35),これらのmiRNAは,それぞれ標的となるがん抑制因子およびがん促進因子の遺伝子発現を抑制することにより,がん促進因子およびがん抑制因子のように振る舞うと考えられている(図3A).
2)がん抑制因子・がん促進因子としてのmiRNA
がんにおけるmiRNAの役割を考える上で,慢性リンパ性白血病(CLL)とmiRNAの関係は欠かすことができない.CLLでは13q14領域の欠失が高頻度で認められるが,2002年,Calin,Croceらのグループは,DLEU2,miR-15a,miR-16-1の三つのncRNAが同部位に存在することを報告した36).CLLでは,miR-15a/miR-16-1の発現低下によってBcl-2の上昇が誘導されアポトーシス抵抗性が惹起されるというモデルが提案された(図3B).これらのmiRNAについては欠損マウスモデルが作製され,病態形成における意義が個体レベルで検証されている37).miR-15a/miR-16-1欠損モデルは,単クローン性B細胞リンパ球増加症,CLL,びまん性大細胞型B細胞性リンパ腫というヒトでのCLLに関連した病態がみられることなどから,ヒトCLLの特徴を忠実に再現しており,悪性腫瘍におけるmiRNAの寄与を示した重要なモデルである.
一方,miRNA研究の初期に同定されたがんに関係する重要な他のmiRNAとして,miR-17-92クラスターがあげられる.びまん性大細胞型B細胞性リンパ腫,濾胞性リンパ腫,マントル細胞リンパ腫などのリンパ腫では13q31領域の増幅がしばしば認められるが,これらの腫瘍では同領域に含まれるmiR-17-92クラスターが腫瘍促進において重要な役割を持っていることが示されている31).
3)がんにおけるmiRNAの発現異常の原因
がんにおけるmiRNAの発現異常の機序として,染色体異常,エピジェネティックな機序,転写レベルでの変化などに加えて,miRNAプロセシングの異常も関与していることが示唆されている(図3C)10,11,38,39).
各がん種におけるmiRNAの発現プロファイルにより腫瘍で発現上昇・発現低下を示すmiRNAがそれぞれ同定されているが,一方で,ヒトの悪性腫瘍ではmiRNAの広範な発現量減少がしばしばみられることが報告されている35,39).これに対応して,DroshaやDicerといったmiRNAプロセシングの中核分子を発現低下させると,細胞の形質転換と腫瘍形成が促進されることが報告されている40).さらに,マウス発がんモデルでの検討により,Dicerのホモ欠損は発がんを抑制するがヘテロ欠損は発がんを促進することから,Dicerはハプロ不全がん抑制因子として機能していることが示唆されており,卵巣がんや肺がんなどでは,Dicerの低発現と予後不良の相関も報告されている41,42).これらの知見は,miRNAによる遺伝子発現調節が細胞の腫瘍化抑制において全体として重要な役割を果たしていることを示唆している.また,がんでは,pre-miRNAの核外輸送を担うXPO5,および,Dicerの補助因子であるTRBP2の遺伝子変異がみられることもある43,44).
1)p53によるmiRNAの発現調節
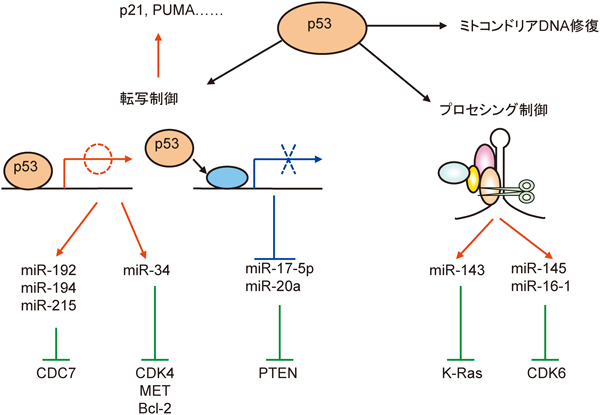
p53は代表的ながん抑制遺伝子であり,多くのヒトがんにおいてp53経路の異常が認められる.DNA損傷に応答してp53はp21,PUMAといったさまざまな標的遺伝子を活性化することにより,細胞周期の停止やアポトーシスを誘導する.p53の中心的な機能は転写活性化因子としての役割でありさまざまなp53の標的遺伝子が同定されているが,p53によって転写制御を受けるmiRNAとしてmiR-34ファミリー,miR-194-2/miR-192,miR-194-1/miR-215クラスター,miR-17-92クラスターなどがある(図4左).miR-34ファミリー(miR-34a, b/c)は,p53によって転写活性化されるmiRNAである45).p53はmiR-34遺伝子群のプロモーターのp53結合配列に結合し,pri-miRNAを誘導することでmiRNAの発現を上昇させる.miR-34はCDK4,E2Fファミリーといった細胞周期関連因子を介してp53の増殖抑制機能に貢献していることが想定されている.
2)p53によるmiRNAプロセシングの制御
p53は転写活性化因子としての機能以外にも,ミトコンドリアにおけるアポトーシスの直接的な制御,DNA修復因子との相互作用によるDNA修復機構の制御などのさまざまな機能を持つことが示されている.これまでに,我々は,p53がDrosha複合体と相互作用し,miRNAプロセシング過程を制御することを見いだしている(図4右)46).
我々は,DNA傷害に伴うmiRNAの発現変化を検討し,miR-34以外に,miR-15/-16/-143/-145などのmiRNAの発現レベルもDNA傷害によって上昇することを見いだした.発生段階やがんにおいて,一次転写産物であるpri-miRNAと成熟型miRNAの間で広範囲な調節が示唆されていることを考慮し39),pri-miRNA,pre-miRNA,成熟型miRNAの発現量を比較した結果,これらのmiRNAについて,pri-miRNAの発現レベルが変化しないにも関わらず,pre-miRNA,成熟型miRNAが上昇することが認められた.この結果は,pri-miRNAの発現制御と独立してmiRNA産生の制御が行われている可能性を示唆するものであった.Drosha複合体はp68/p72と呼ばれるDEAD-box型RNAヘリカーゼなどの補助因子を伴うが,一方で,p68/p72はさまざまな転写因子のコファクターとして働くことも知られている.検討の結果,DNA損傷に応答して,p53がp68/p72依存性にDrosha複合体と相互作用し,Droshaのプロセシング機能を促進することが明らかとなった.また,悪性腫瘍でみられる変異型p53の一部が野生型p53とは逆にmiRNAのプロセシングを阻害することも見いだされた.miR-16/-143といったDNA傷害によって誘導されるmiRNAは増殖抑制機能を持っており,これらのmiRNA群もp53の増殖抑制機能を補完していると考えられる38).また,最近の報告により,p53の120番目のリシンのアセチル化がDroshaとの会合を促進し,pri-miRNAのプロセシングを促進することも報告されている47).
miRNAは遺伝子発現のファインチューナーと称されるが,miRNAの生合成自体もファインチューニングを受けていることが次々と報告されている(図5)5,48).TGF-β(transforming growth factor-β)シグナル伝達を担うSmadはp53と類似のメカニズムでmiRNAのプロセシングを制御する49).さらに,Lin28,hnRNP A1,KSRPといったさまざまなRNA結合タンパク質が特定のmiRNAのプロセシングを制御することも報告されている.Lin28は,がん抑制因子であるlet-7のプロセシングを,Drosha,Dicerレベルで特異的に阻害する.Lin28はLin28AとLin28Bの二つの遺伝子が知られているが,Lin28Aは,pre-let-7へ結合することにより,TUT4によるpre-let-7の3′末端のポリウリジニル化,およびDis3l2エキソヌクレアーゼによるpre-let-7の分解を誘導しプロセシングを阻害する50–52).一方で,Lin28BはLin28Aと異なり主に核内でpri-let-7のプロセシングを抑制することが報告されているが,Lin28AとLin28Bは同様の生化学的特徴を持っていることもあり,そのメカニズムの差異については議論の余地が残っている53).また,RNAエディティングによるmiRNA前駆体の修飾や,EGFRによるAgo2のリン酸化などを介して,miRNAの生合成や活性が調節されることも報告されている.
7. MCPIP1によるmiRNA生合成の負の制御
RNAi機構/miRNA生合成機構に関係する分子群は,多くの生物種で幅広く保存されている.RNAiは植物や無脊椎動物において主要な抗ウイルス応答として機能するが,哺乳類では,インターフェロン経路が非常に強力な抗ウイルス応答として存在しており,RNAi機構の生理的意義における進化的変遷がうかがえる.また,miRNAを産生する過程の調節機構に比べて,miRNA前駆体・成熟型miRNAのターンオーバーの調節機構については不明な点が多い.
続いて,我々は,哺乳類において,免疫応答に関係し,またRNA結合ドメインを有する遺伝子がmiRNA生合成の調節に関与する可能性を検討した.結果として,免疫応答の制御に関わるMCPIP1(Zc3h12a)という分子が,miRNAの活性および生合成を強力に抑制することを見いだした(図6)54).検討の結果,MCPIP1は,細胞質でエンドリボヌクレアーゼとしてpre-miRNAのターミナルループ部分を切断・分解することにより,Dicerと拮抗しmiRNAの生合成を阻害することが明らかとなった.肺がんなどではDicerの発現低下が予後不良と関係することが報告されているが,肺がんのトランスクリプトーム解析により,MCPIP1とDicerが拮抗関係にあり,Dicerとは逆にMCPIP1の高発現と予後不良が相関することも見いだされた.MCPIP1は炎症応答によって発現調節を受ける分子であり,MCPIP1は炎症応答とがんにおけるmiRNA生合成機能不全を結びつける分子といえるかもしれない55).
8. がんにおけるmiRNAの多面的な役割:がん微小環境の制御
従来の研究では,個々のmiRNAの,細胞周期,細胞死,転移能といったがん細胞の特性そのものに与える影響(細胞自律的な機能)に焦点がおかれていたことが多かったが,近年,miRNAの細胞非自律的な機能にも注目が集まっており,がん微小環境や血管新生におけるmiRNAの意外な働きも明らかになってきている56).がんで発現低下がみられるmiR-126について,miR-126ががん細胞から分泌される血管新生促進因子を抑制することにより,血管新生と転移を抑制することが示されている57).また,我々も,NPM-ALK陽性悪性リンパ腫(未分化大細胞型リンパ腫)で高発現するmiR-135bがGAT A3やSTAT6といったTh2細胞分化のマスター因子を抑制することにより,リンパ腫細胞をTh17細胞様の免疫形質に偏向させ,炎症性サイトカインの分泌亢進・がん微小環境における炎症応答の促進を誘導することを見いだしている58).さらに,miRNAは,より直接的に細胞間のコミュニケーションで重要な役割を持っていることが明らかになっており,特に,エキソソームに含まれた分泌型miRNAは大きな注目を集めている59).
1)mRNA/miRNAのペアリング統合解析
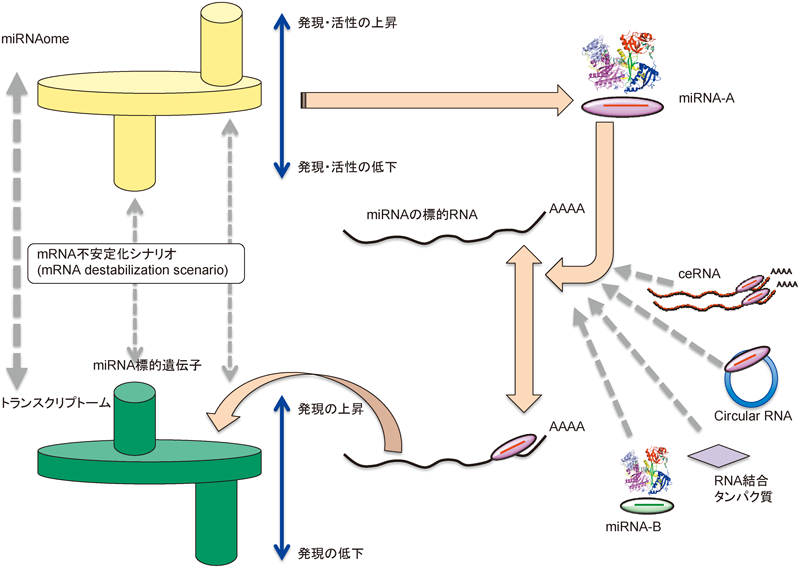
miRNAによる遺伝子抑制は,配列相補性以外に,seed領域付近のAU塩基の割合,3′UTR内における標的配列の位置などによって影響される4,23,60).定量的解析により,miRNAが主に標的mRNAの発現量を低下させることでタンパク質の発現を抑制していることが提案されている(mRNA destabilization scenario).このことは,mRNAの発現プロファイルが,対応するmiRNAの発現または活性の変化を反映していることを示唆するものである(図7)27–30).しかし,mRNA destabilization scenarioは一つのmiRNAを多量に発現,あるいは強く抑制した場合,または少数の組織特異的なmiRNAに注目した場合にみられる現象であり,複数のmiRNAが小さな変動幅で変化するような疾患のトランスクリプトームの解析で果たして同様に適用できるかどうかは不明であった.
我々は,mRNAプロファイリングのデータからmiRNAの活性を推定するアプローチを構築し,その結果をペアとなるmiRNAプロファイリングのデータと比較することでmRNA destabilization scenarioが現実的な状況でも広く適用可能であるかどうかを検証した61,62).結果として,Gene Set Enrichment Analysis(GSEA)と,遺伝子セット内のmiRNA標的遺伝子のenrichmentを評価する手法(Functional Assignment of miRNAs via Enrichment: FAME)を組み合わせる手法を考案した(GSEA-FAME analysis: GFA).悪性リンパ腫における解析で,GFAと他の手法を比較した結果,GFAにより,miRNAの発現パターンと,mRNAプロファイルから推定されるmiRNAの活性パターンの間により強い相関を見いだすことができた.これは,mRNA destabilization scenarioの現実的な局面における実証といえる.
2)mRNA/miRNA統合解析の有用性
mRNAプロファイリングやmiRNAプロファイリングといった網羅的解析の大きな目的の一つは,多数の情報から対象となる生物学的過程をより特徴づける情報を抽出することにあるといえる.がんの分野では,特定の遺伝子シグネチャーを抽出し,これを用いてがんの分類や予後予測を行うことが盛んに行われている.mRNA/miRNAのペアリング解析において,miRNAのプロファイリングと活性予測の双方で大きな変動を示すマーカーが,より頑健なバイオマーカーとして機能する可能性を検討し,発現量のプロファイリングに加えてmiRNA–mRNAネットワークの関係性を考慮することで,より頑健なバイオマーカーをゲノムワイド発現解析から抽出できる可能性を見いだしている62).
10. More read, More mystery
1)miRNAとは何か:miRNA生合成の謎
近年の次世代シーケンサーによる低分子RNA解析は,成熟を遂げつつあるmiRNA研究に,新しい問いを生み出している.これらの解析により,新しいmiRNAが同定されるとともに,生体内にあるmiRNAの不均一性(isomiRの存在,塩基付加・欠失)が明らかになっている.また,RISCの形成では,miRNA duplexからガイド鎖が最終的にAgoタンパク質に残り,もう一方の鎖(miRNA*鎖,パッセンジャー鎖)はAgoタンパク質より取り除かれる.従来的には,5′末端がより不安定な側の鎖がRISCに取り込まれ(熱力学的安定性ルール),miRNA鎖とmiRNA*鎖のバイアスが生じることになるが,miRNA鎖,miRNA*鎖の定義は絶対的なものではなく,miRNA*鎖と定義されているものもかなりの割合でRISCに取り込まれ機能する場合があることがわかっており,次世代シーケンサーによる解析はこれをはっきり支持している63,64).ガイド鎖とパッセンジャー鎖を決定づける分子生物学的ルールとメカニズムは何なのか.この古くて新しい謎は,miRNAとは何か,二本鎖RNAを起点とするRNA干渉でどちらのRNAが機能するか,という根源的な問題と直結しており,核酸医薬の最適化を考える上でも重要である.
2)miRNA regulon:発現と機能の間に
さまざまなアプローチでmiRNA/mRNAプロファイリングを解析することはmiRNAの機能を明らかにする上で重要であるが,一方で,miRNAの産生からmiRNAが実際に機能するまでの間に,さまざまな調節機構が介在してmiRNA/RNA結合タンパク質/標的mRNA間のネットワークを形成していることが明らかになりつつある(図7).competing endogenous RNA(ceRNA)コンセプトやcircular RNAなどに代表されるRNA間でのクロストークが現在注目を集めている.ceRNAについては,miRNAとmRNAのコピー数の相対比を考慮した際に実際にそのような現象が起こっているのかについて議論が盛んに行われているが65–67),転写後調節レベルで,miRNAと他のmiRNAの間で同様の現象が起こっている可能性は十分にあるだろう.
本稿では,miRNAの生合成調節機構と遺伝子発現調節機構に関するこれまでの知見を紹介した.RNA干渉の治療応用にはドラッグデリバリーの問題などいまだ解決すべき問題点が多いが,近年,miRNAの導入や抑制による治療応用の可能性が報告されつつある68).miRNAの生合成のさらなる理解は治療応用の可能性を拡大するために必須であり,特に,前述した低分子RNAの非対称性の問題については,今後の研究の発展が期待される.また,miRNA–mRNAネットワークの解析は,遺伝子発現におけるファインチューナーという言葉だけでは語りきれないmiRNAの新しい生物学的意義もさらに明らかにしてゆく可能性を秘めている.
謝辞Acknowledgments
本稿で紹介した筆者らの研究成果は,東京大学大学院医学系研究科分子病理学教室で,宮園浩平先生の指導のもと行われたものです.宮園浩平先生,共同研究者の方々,そして,現所属のPhillip A. Sharp先生にこの場を借りて深く感謝致します.
引用文献References
1) Lee, R.C., Feinbaum, R.L., & Ambros, V. (1993) Cell, 75, 843–854.
2) Wightman, B., Ha, I., & Ruvkun, G. (1993) Cell, 75, 855–862.
3) Fire, A., Xu, S., Montgomery, M.K., Kostas, S.A., Driver, S.E., & Mello, C.C. (1998) Nature, 391, 806–811.
4) Bartel, D.P. (2004) Cell, 116, 281–297.
5) Ha, M. & Kim, V.N. (2014) Nat. Rev. Mol. Cell Biol., 15, 509–524.
6) Lagos-Quintana, M., Rauhut, R., Lendeckel, W., & Tuschl, T. (2001) Science, 294, 853–858.
7) Lau, N.C., Lim, L.P., Weinstein, E.G., & Bartel, D.P. (2001) Science, 294, 858–862.
8) Lee, R.C. & Ambros, V. (2001) Science, 294, 862–864.
9) Calin, G.A. & Croce, C.M. (2006) Nat. Rev. Cancer, 6, 857–866.
10) Croce, C.M. (2009) Nat. Rev. Genet., 10, 704–714.
11) Suzuki, H.I. & Miyazono, K. (2011) J. Biochem., 149, 15–25.
12) Denli, A.M., Tops, B.B., Plasterk, R.H., Ketting, R.F., & Hannon, G.J. (2004) Nature, 432, 231–235.
13) Gregory, R.I., Yan, K.P., Amuthan, G., Chendrimada, T., Doratotaj, B., Cooch, N., & Shiekhattar, R. (2004) Nature, 432, 235–240.
14) Auyeung, V.C., Ulitsky, I., McGeary, S.E., & Bartel, D.P. (2013) Cell, 152, 844–858.
15) Liu, J., Carmell, M.A., Rivas, F.V., Marsden, C.G., Thomson, J.M., Song, J.J., Hammond, S.M., Joshua-Tor, L., & Hannon, G.J. (2004) Science, 305, 1437–1441.
16) Kim, V.N. (2005) Nat. Rev. Mol. Cell Biol., 6, 376–385.
17) Zamudio, J.R., Kelly, T.J., & Sharp, P.A. (2014) Cell, 156, 920–934.
18) Su, H., Trombly, M.I., Chen, J., & Wang, X. (2009) Genes Dev., 23, 304–317.
19) Schurmann, N., Trabuco, L.G., Bender, C., Russell, R.B., & Grimm, D. (2013) Nat. Struct. Mol. Biol., 20, 818–826.
20) Hauptmann, J., Dueck, A., Harlander, S., Pfaff, J., Merkl, R., & Meister, G. (2013) Nat. Struct. Mol. Biol., 20, 814–817.
21) Faehnle, C.R., Elkayam, E., Haase, A.D., Hannon, G.J., & Joshua-Tor, L. (2013) Cell Reports, 3, 1901–1909.
22) Nakanishi, K., Ascano, M., Gogakos, T., Ishibe-Murakami, S., Serganov, A.A., Briskin, D., Morozov, P., Tuschl, T., & Patel, D.J. (2013) Cell Reports, 3, 1893–1900.
23) Grimson, A., Farh, K.K., Johnston, W.K., Garrett-Engele, P., Lim, L.P., & Bartel, D.P. (2007) Mol. Cell, 27, 91–105.
24) Tay, Y., Zhang, J., Thomson, A.M., Lim, B., & Rigoutsos, I. (2008) Nature, 455, 1124–1128.
25) Loeb, G.B., Khan, A.A., Canner, D., Hiatt, J.B., Shendure, J., Darnell, R.B., Leslie, C.S., & Rudensky, A.Y. (2012) Mol. Cell, 48, 760–770.
26) Helwak, A., Kudla, G., Dudnakova, T., & Tollervey, D. (2013) Cell, 153, 654–665.
27) Lim, L.P., Lau, N.C., Garrett-Engele, P., Grimson, A., Schelter, J.M., Castle, J., Bartel, D.P., Linsley, P.S., & Johnson, J.M. (2005) Nature, 433, 769–773.
28) Baek, D., Villen, J., Shin, C., Camargo, F.D., Gygi, S.P., & Bartel, D.P. (2008) Nature, 455, 64–71.
29) Guo, H., Ingolia, N.T., Weissman, J.S., & Bartel, D.P. (2010) Nature, 466, 835–840.
30) Subtelny, A.O., Eichhorn, S.W., Chen, G.R., Sive, H., & Bartel, D.P. (2014) Nature, 508, 66–71.
31) He, L., Thomson, J.M., Hemann, M.T., Hernando-Monge, E., Mu, D., Goodson, S., Powers, S., Cordon-Cardo, C., Lowe, S.W., Hannon, G.J., & Hammond, S.M. (2005) Nature, 435, 828–833.
32) O’Donnell, K.A., Wentzel, E.A., Zeller, K.I., Dang, C.V., & Mendell, J.T. (2005) Nature, 435, 839–843.
33) Johnson, S.M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K.L., Brown, D., & Slack, F.J. (2005) Cell, 120, 635–647.
34) Calin, G.A., Ferracin, M., Cimmino, A., Di Leva, G., Shimizu, M., Wojcik, S.E., Iorio, M.V., Visone, R., Sever, N.I., Fabbri, M., Iuliano, R., Palumbo, T., Pichiorri, F., Roldo, C., Garzon, R., Sevignani, C., Rassenti, L., Alder, H., Volinia, S., Liu, C.G., Kipps, T.J., Negrini, M., & Croce, C.M. (2005) N. Engl. J. Med., 353, 1793–1801.
35) Lu, J., Getz, G., Miska, E.A., Alvarez-Saavedra, E., Lamb, J., Peck, D., Sweet-Cordero, A., Ebert, B.L., Mak, R.H., Ferrando, A.A., Downing, J.R., Jacks, T., Horvitz, H.R., & Golub, T.R. (2005) Nature, 435, 834–838.
36) Calin, G.A., Dumitru, C.D., Shimizu, M., Bichi, R., Zupo, S., Noch, E., Aldler, H., Rattan, S., Keating, M., Rai, K., Rassenti, L., Kipps, T., Negrini, M., Bullrich, F., & Croce, C.M. (2002) Proc. Natl. Acad. Sci. USA, 99, 15524–15529.
37) Klein, U., Lia, M., Crespo, M., Siegel, R., Shen, Q., Mo, T., Ambesi-Impiombato, A., Califano, A., Migliazza, A., Bhagat, G., & Dalla-Favera, R. (2010) Cancer Cell, 17, 28–40.
38) Suzuki, H.I. & Miyazono, K. (2010) J. Mol. Med (Berl), 88, 1085–1094.
39) Thomson, J.M., Newman, M., Parker, J.S., Morin-Kensicki, E.M., Wright, T., & Hammond, S.M. (2006) Genes Dev., 20, 2202–2207.
40) Kumar, M.S., Lu, J., Mercer, K.L., Golub, T.R., & Jacks, T. (2007) Nat. Genet., 39, 673–677.
41) Kumar, M.S., Pester, R.E., Chen, C.Y., Lane, K., Chin, C., Lu, J., Kirsch, D.G., Golub, T.R., & Jacks, T. (2009) Genes Dev., 23, 2700–2704.
42) Merritt, W.M., Lin, Y.G., Han, L.Y., Kamat, A.A., Spannuth, W.A., Schmandt, R., Urbauer, D., Pennacchio, L.A., Cheng, J.F., Nick, A.M., Deavers, M.T., Mourad-Zeidan, A., Wang, H., Mueller, P., Lenburg, M.E., Gray, J.W., Mok, S., Birrer, M.J., Lopez-Berestein, G., Coleman, R.L., Bar-Eli, M., & Sood, A.K. (2008) N. Engl. J. Med., 359, 2641–2650.
43) Melo, S.A., Ropero, S., Moutinho, C., Aaltonen, L.A., Yamamoto, H., Calin, G.A., Rossi, S., Fernandez, A.F., Carneiro, F., Oliveira, C., Ferreira, B., Liu, C.G., Villanueva, A., Capella, G., Schwartz, S. Jr., Shiekhattar, R., & Esteller, M. (2009) Nat. Genet., 41, 365–370.
44) Melo, S.A., Moutinho, C., Ropero, S., Calin, G.A., Rossi, S., Spizzo, R., Fernandez, A.F., Davalos, V., Villanueva, A., Montoya, G., Yamamoto, H., Schwartz, S. Jr., & Esteller, M. (2010) Cancer Cell, 18, 303–315.
45) He, L., He, X., Lim, L.P., de Stanchina, E., Xuan, Z., Liang, Y., Xue, W., Zender, L., Magnus, J., Ridzon, D., Jackson, A.L., Linsley, P.S., Chen, C., Lowe, S.W., Cleary, M.A., & Hannon, G.J. (2007) Nature, 447, 1130–1134.
46) Suzuki, H.I., Yamagata, K., Sugimoto, K., Iwamoto, T., Kato, S., & Miyazono, K. (2009) Nature, 460, 529–533.
47) Chang, J., Davis-Dusenbery, B.N., Kashima, R., Jiang, X., Marathe, N., Sessa, R., Louie, J., Gu, W., Lagna, G., & Hata, A. (2013) EMBO J., 32, 3192–3205.
48) Siomi, H. & Siomi, M.C. (2010) Mol. Cell, 38, 323–332.
49) Davis, B.N., Hilyard, A.C., Lagna, G., & Hata, A. (2008) Nature, 454, 56–61.
50) Heo, I., Joo, C., Cho, J., Ha, M., Han, J., & Kim, V.N. (2008) Mol. Cell, 32, 276–284.
51) Heo, I., Joo, C., Kim, Y.K., Ha, M., Yoon, M.J., Cho, J., Yeom, K.H., Han, J., & Kim, V.N. (2009) Cell, 138, 696–708.
52) Chang, H.M., Triboulet, R., Thornton, J.E., & Gregory, R.I. (2013) Nature, 497, 244–248.
53) Piskounova, E., Polytarchou, C., Thornton, J.E., LaPierre, R.J., Pothoulakis, C., Hagan, J.P., Iliopoulos, D., & Gregory, R.I. (2011) Cell, 147, 1066–1079.
54) Suzuki, H.I., Arase, M., Matsuyama, H., Choi, Y.L., Ueno, T., Mano, H., Sugimoto, K., & Miyazono, K. (2011) Mol. Cell, 44, 424–436.
55) Suzuki, H.I. & Miyazono, K. (2012) The Enzymes, 32, 163–183.
56) Suzuki, H.I., Katsura, A., Matsuyama, H., & Miyazono, K. (2014) Oncogene. 10.1038/onc.2014.254
57) Png, K.J., Halberg, N., Yoshida, M., & Tavazoie, S.F. (2012) Nature, 481, 190–194.
58) Matsuyama, H., Suzuki, H.I., Nishimori, H., Noguchi, M., Yao, T., Komatsu, N., Mano, H., Sugimoto, K., & Miyazono, K. (2011) Blood, 118, 6881–6892.
59) Kosaka, N., Iguchi, H., & Ochiya, T. (2010) Cancer Sci., 101, 2087–2092.
60) Lewis, B.P., Burge, C.B., & Bartel, D.P. (2005) Cell, 120, 15–20.
61) Suzuki, H.I., Matsuyama, H., Noguchi, M., Yao, T., Komatsu, N., Mano, H., Sugimoto, K., & Miyazono, K. (2013) Leukemia, 27, 2107–2111.
62) Suzuki, H.I., Mihira, H., Watabe, T., Sugimoto, K., & Miyazono, K. (2013) Nucleic Acids Res., 41, e62.
63) Chiang, H.R., Schoenfeld, L.W., Ruby, J.G., Auyeung, V.C., Spies, N., Baek, D., Johnston, W.K., Russ, C., Luo, S., Babiarz, J.E., Blelloch, R., Schroth, G.P., Nusbaum, C., & Bartel, D.P. (2010) Genes Dev., 24, 992–1009.
64) Yang, J.S., Phillips, M.D., Betel, D., Mu, P., Ventura, A., Siepel, A.C., Chen, K.C., & Lai, E.C. (2011) RNA, 17, 312–326.
65) Salmena, L., Poliseno, L., Tay, Y., Kats, L., & Pandolfi, P.P. (2011) Cell, 146, 353–358.
66) Denzler, R., Agarwal, V., Stefano, J., Bartel, D.P., & Stoffel, M. (2014) Mol. Cell, 54, 766–776.
67) Bosson, A.D., Zamudio, J.R., & Sharp, P.A. (2014) Mol. Cell, 56, 347–359.
68) Janssen, H.L., Reesink, H.W., Lawitz, E.J., Zeuzem, S., Rodriguez-Torres, M., Patel, K., van der Meer, A.J., Patick, A.K., Chen, A., Zhou, Y., Persson, R., King, B.D., Kauppinen, S., Levin, A.A., & Hodges, M.R. (2013) N. Engl. J. Med., 368, 1685–1694.
著者紹介Author Profile
 鈴木 洋(すずき ひろし)
鈴木 洋(すずき ひろし)マサチューセッツ工科大学コーク癌総合研究所客員研究員.医学博士.
略歴1979年大阪府生まれ,愛媛県育ち.2004年東京大学医学部医学科卒業.3年の臨床研修を経て,10年同大学院医学系研究科早期修了(宮園浩平教授),分子病理学分野特任助教.14年より現所属(Phillip A. Sharp教授).
研究テーマと抱負RNAバイオロジーに焦点をおいた,遺伝子発現調節ネットワークの理解,悪性腫瘍などの分子病態の理解,新規治療法の開拓.Sciences for precision medicineを目指している.