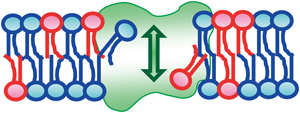
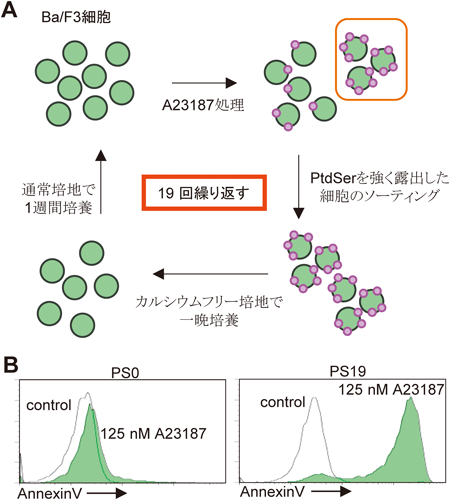
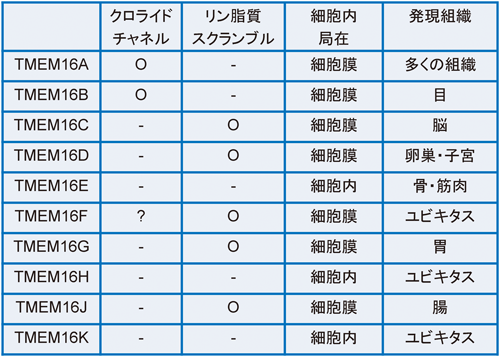
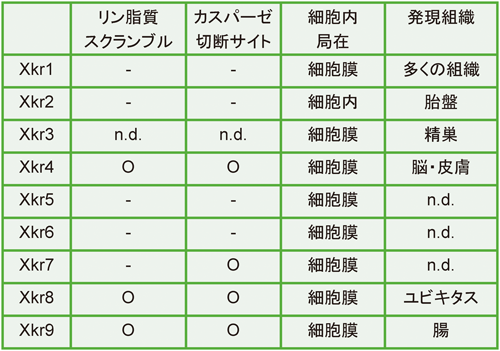
細胞膜リン脂質のスクランブル機構Mechanisms of phospholipid scrambling on plasma membrane
大阪大学免疫学フロンティア研究センター免疫グループ免疫・生化学研究室Laboratory of Biochemistry and Immunology, Immunology Group, Immunology Frontier Research Center, Osaka University ◇ 〒565-0871 大阪府吹田市山田丘3番1号3-1 Yamadaoka, Suita-shi, Osaka 565-0871, Japan
発行日:2015年8月25日Published: August 25, 2015