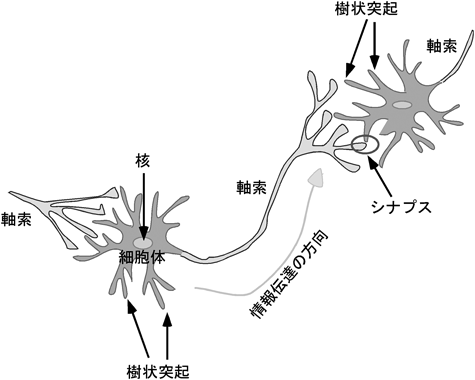
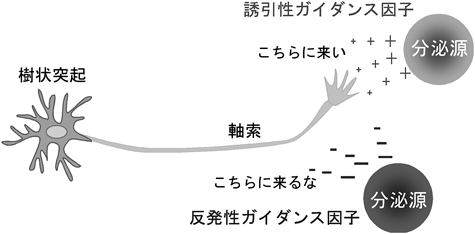
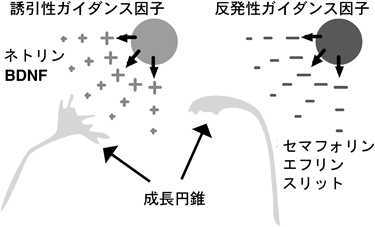
学習や記憶といった高次脳機能を可能としているのは,神経細胞が神経突起を伸長し,互いにシナプスを築くことによって形成される複雑な神経回路である.数多くの先人の解剖学的な研究により,多くの脳神経回路のパターンがわかっていた.神経細胞先端には成長円錐という構造体があり,この構造体が周辺のガイダンス因子を感知して軸索を的確な方向へ伸長させている.今から100年以上前の1892年にスペインの解剖学者Ramón y Cajalは発生期の軸索先端に扇状の膨大部(成長円錐)があることを発見し,これが周囲の微小環境に存在する何らかの手がかりを感知して軸索の伸長方向を決定しているという考え(化学向性説:chemotropism)をすでに提唱していた1).しかし,それらの形成機構に関する分子レベルの研究が手がけられたのは,ここ10年来のことである.脳神経系の構成単位である神経細胞は特異的な極性を持ち,通常は1本の長い軸索と複数の複雑に分枝した樹状突起を細胞体から伸展している(図1).神経細胞間の情報伝達は軸索と樹状突起で構築されるシナプスを介して行われ,樹状突起や細胞体で受け取った情報は細胞体に集約され,軸索を通って隣の神経細胞の樹状突起や細胞体へ神経伝達物質が放出されることによって伝わる.複雑で正確な神経回路網を形成するために,神経細胞はさまざまな制御を受けてその形態がかたち作られており,その中の一つにガイダンス因子による誘導作用がある.軸索ガイダンス因子は,誘引作用(こちらの方向に伸びて来い)を示す誘引性ガイダンス因子と,反発作用(こちらの方向に伸びて来るな)を示す反発性ガイダンス因子とに大きく分けられ,誘引作用を示すガイダンス因子の方向に神経軸索は伸長していき,反発作用を示すガイダンス因子を遠ざけるようにして伸長する(図2).軸索ガイダンス分子にはさまざまな分子種があり,反発作用を示す分子にはセマフォリン(semaphorin)ファミリー,エフリン(ephrin)ファミリー,slitファミリーなどが知られており,誘引作用を示す分子にはネトリン(netrin)や脳由来神経栄養因子(brain-derived neurotrophic factor: BDNF)などの神経成長因子などが知られている(図3).
筆者らは,代表的な反発性ガイダンス因子であるセマフォリンの細胞内情報伝達機構の研究を行ってきた.セマフォリンは元来,発達期の神経軸索伸長における忌避物質,「反発性神経軸索ガイダンス因子」として同定された分子である.近年,その機能は神経系にとどまらず,心血管系形成2,3),骨代謝4),免疫シナプス形成5),がん転移抑制6,7)など,生体形成のさまざまな場面において,さらには,生理,病態のさまざまな場面において細胞のナビゲーションを担っており,いわば,「生体形成ガイダンス因子」としてきわめて重要なタンパク質であることが明らかになってきている.筆者らは,そのセマフォリンの受容体,プレキシン(plexin)の細胞内情報伝達経路の解明に取り組み,膜1回貫通型受容体であるプレキシンの細胞内領域そのものがRasファミリー低分子量Gタンパク質,R-Rasに対する直接の不活性化因子,GAP(GTPase-activating protein)として働き,神経軸索の伸長8)や細胞運動の亢進9,10)に寄与するR-Rasの働きにブレーキをかけるという,新奇な情報伝達機構を明らかにした11).以来,その情報伝達機構が神経系に限らず,幅広い細胞の細胞運動や形態の制御において普遍的に駆動されるシステムであることが明らかになり,さらには,セマフォリン以外の他のガイダンス因子のガイダンスにも,R-Ras活性変化を伴うことがわかってきた.本稿では,低分子量Gタンパク質,R-Rasを窓とし,さまざまな細胞種におけるガイダンス機能発揮の分子基盤となる一連の細胞形態調節のシグナル伝達経路に関して紹介する.
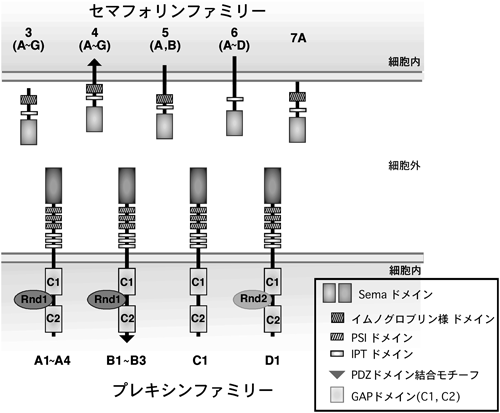
2. セマフォリン受容体,プレキシンの細胞内領域内の保存されたドメインの役割
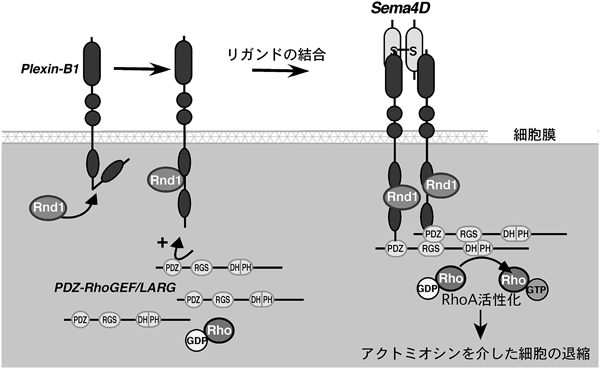
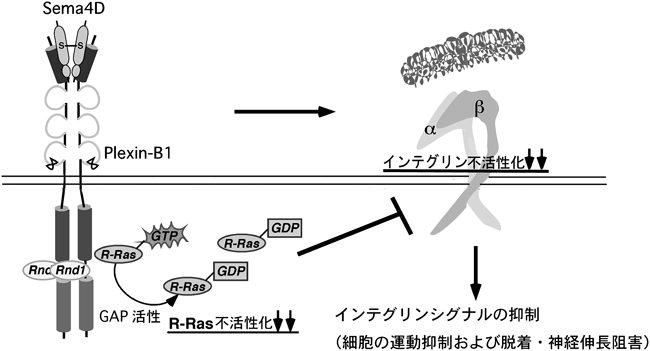
解剖学的研究が主流だった時代から,神経回路形成において,何らかの軸索誘導因子の存在は想定されていたのだが,その実体は長らく明らかではなかった.その後,分子レベルの研究がなされるようになり,1993年にニワトリの脳からコラプシン(collapsin)という軸索ガイダンス因子が分離,精製されて,初めてその実態が明らかとなった12).このコラプシンが現在セマフォリン3A(Sema3A)と呼ばれる分子であり,その後に多くのセマフォリンファミリーのメンバーが見つかっていった.セマフォリンファミリーは,線虫からヒトまで種を超えて幅広く存在する.現在までに8クラスが同定されており,分泌型(Sema2, Sema3, SemaV),細胞膜1回貫通型(Sema1, Sema4, Sema5, Sema6)やGPIアンカー型(Sema7)が存在する.セマフォリンの受容体として,ニューロピリン(neuropilin)とプレキシンが同定されているが,ニューロピリンは細胞内領域がきわめて短く,セマフォリンによる情報伝達はプレキシンを介して行われる.プレキシンはAからDまでの四つのサブファミリーに分類され,Plexin-A1~4,Plexin-B1-3,Plexin-C1,そしてPlexin-D1からなっている13)(図4).哺乳類を含めた脊椎動物のPlexin-Bサブファミリーに限って,カルボキシ末端にはPDZ(PSD-95/Dlg/ZO-1)ドメイン結合モチーフが存在する.複数グループの報告により,このドメインは低分子量Gタンパク質,RhoAの活性化を担っていることが,同時期に次々に明らかにされた14~17).Plexin-Bサブファミリーのカルボキシ末端のPDZドメイン結合モチーフに,PDZドメインを保有するRhoAに対する活性化因子,グアニンヌクレオチド交換因子(guanine nucleotide exchange factor: GEF)であるPDZ-RhoGEFやLARG(leukemia-associated Rho GEF)が直接結合することで,Plexin-Bサブファミリーは細胞内でRhoAを活性化し,下流のエフェクターであるRhoキナーゼを介したアクトミオシン収縮を引き起こすことによって,細胞の退縮や神経突起退縮をはじめとする反発性作用を発揮する.筆者らも,Plexin-B1がリガンドであるSema4D依存的なRhoA活性化を介した細胞の退縮応答を引き起こすメカニズムを明らかにした.具体的な機序は図5に示したとおりである.Plexin-B1の細胞内領域にはRhoファミリー低分子量Gタンパク質の一つ,Rnd1が直接結合する.このRnd1が結合することが,Plexin-B1とPDZ-RhoGEFとの結合を促進し,リガンドであるSema4D依存的なRhoA活性化を増強するというシステムを,筆者らはまず明らかにした18).
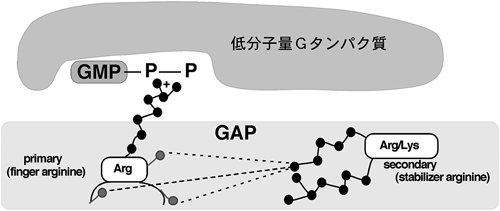
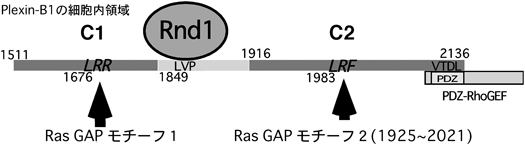
しかしながら,PDZ結合モチーフが,Bサブファミリー以外のプレキシンには存在しないことから,筆者らは,プレキシンの反発性応答の要となる機能ドメインは他にあると考えた.プレキシンファミリーの細胞内領域には,サブファミリーや動物種を超えてよく保存されている二つの領域,C1領域とC2領域があるが,この保存された領域がどのような機能を発現しているのかはまったく不明であった.セマフォリンは元来,発達期の神経系においての反発因子として働く分子として単離されたが12),その後の報告で,セマフォリンが神経系以外を含めた幅広い細胞種で細胞接着や細胞運動を制御していることがわかってきた19~23).C1領域およびC2領域のアミノ酸配列は,低分子量Gタンパク質,特にRasファミリーの活性抑制分子,GAPと低い相同性があることが,以前から知られていた20,24).しかし,この保存された領域が実際に発揮する機能はまったく不明であった.低分子量Gタンパク質のさまざまなGAPの結晶構造解析が行われ,低分子量Gタンパク質とGAPの結合や,GAP活性の発現の様式が明らかにされている25).一般に,低分子量Gタンパク質に対するGAPタンパク質の分子内には,よく保存されているアルギニン残基を含むArgモチーフが二つ存在する.これらprimary Argモチーフおよびsecondary Argモチーフが分子内で立体的なブリッジを形成し,そのポケットに低分子量Gタンパク質が結合し,そのGTPase活性が促進される(図6).筆者らがPlexin-B1の細胞内領域の配列を詳細に検討したところ,primary ArgモチーフがC1領域に,secondary ArgモチーフがC2領域に存在した.さらに,Plexin-B1に対するRnd1の結合領域は,C1領域とC2領域の間のリンカー部分に位置することがわかった(図7).このことから,Rnd1の結合がGAP活性の発現に重要な役割を果たしうると考えた.次に,プレキシンがRasファミリーに対するGAPとして働くのであれば,どのRasに対するGAPであるかを推定した.プレキシンが反発作用を示すという事実から,R-Rasに対するGAPとして働くのではないかと考えた.R-RasはRasファミリーに属する低分子量Gタンパク質で,細胞接着因子受容体であるβ1インテグリン(β1 integrin)を活性化して細胞膜の伸展や神経突起伸長,細胞運動などに関わっている8–10,26).Plexin-B1のR-Rasに対するGAP活性を調べたところ,Plexin-B1はRnd1の結合依存的に,in vitroおよびin vivoでSema4D刺激依存的にR-Rasに対するGAP活性を示した.しかも,Plexin-B1のGAP活性はR-Rasに選択性があり,別の代表的なRasファミリー低分子量Gタンパク質であるH-Rasに対するGAP活性はなかった.Plexin-B1のC1領域,C2領域内のprimaryおよびsecondary Argモチーフに変異を入れたPlexin-B1-RA(R1677A, R1678A, R1984A)の発現ではSema4Dによる神経突起退縮が起こらなかったことから,このPlexin-B1のR-Ras GAP活性が神経細胞の反発作用に必要であることが示された.また,恒常的活性型のR-RasであるR-RasQL(Q87L)を導入した細胞や,RNA干渉により細胞内在性のRnd1の発現を抑制した細胞では,Sema4Dによる神経突起に対する反発作用が阻害された.さらに,Rnd1はPlexin-A1の細胞内領域にも結合することが知られていたので20,27),Sema3Aの下流シグナルにおいてもPlexin-A1がR-Ras GAPとして働くことが反発性作用に必要か検討したところ,初代培養神経細胞におけるSema3Aによる成長円錐崩壊もR-RasQLの導入により阻止された.また,筆者らや他のグループのその後の研究で,Plexin-CおよびPlexin-Dを含め,他のすべてのプレキシンサブファミリーがR-Rasに対するGAPとして働くことが示された28,29).以上の結果から,プレキシンの細胞内領域自体はR-Ras GAPとして働き,さらにその活性はプレキシンファミリーに普遍的なシグナル伝達経路であることが示唆される.
3. プレキシンによるインテグリン活性抑制のシグナル伝達経路
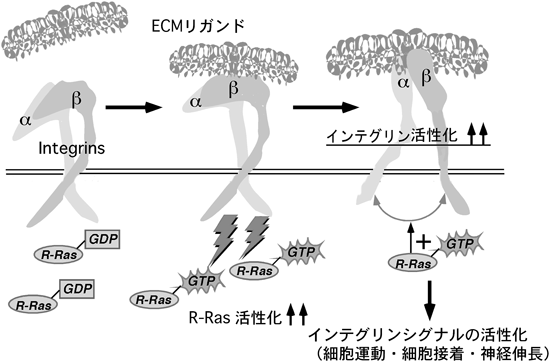
近年の報告で,セマフォリンが神経系以外を含めた幅広い細胞種で細胞接着や細胞運動を制御していることがわかってきた19–23).たとえば,Sema4DによるPlexin-B1の活性化は線維芽細胞において,インテグリンを介した細胞接着を抑制する30).また,別のプレキシンサブファミリーのPlexin-C1は樹状細胞においてケモカインによって引き起こされる細胞運動を抑制する31).しかし,どのような分子機構でセマフォリンが幅広い細胞種の細胞接着や細胞運動を制御しているかは謎であった.R-Rasは細胞接着因子受容体インテグリンを活性化することで細胞接着を促進し,細胞運動や神経突起伸長を引き起こす8,10,26).筆者らはこれらから,Sema4D/Plexin-B1によって発揮されるR-Ras GAP活性が,R-Rasの不活性化を引き起こし,インテグリンの活性化の抑制,細胞と基質の接着の低下をもたらし,細胞運動の抑制を引き起こすと推測した.インテグリンはα,βからなるヘテロ二量体として存在する細胞膜表面受容体で,コラーゲンやフィブロネクチンといった細胞外基質(extracellular matrix: ECM)に結合し,細胞の増殖,生存,運動,そして,がん細胞の転移に重要な役割を担っている32).インテグリンの活性化は細胞接着,運動に必要であり,これまでの多くの研究によって,Rasファミリーに属する低分子量Gタンパク質がインテグリンの活性を制御していることがわかっている10).その中で,活性型のR-Rasはインテグリンの立体的活性化構造を引き起こし,インテグリンをよりECMとの結合能が高い立体構造状態にすることで細胞接着を亢進することが知られており,R-Rasがインテグリンの活性制御において重要な役割を果たしていることが示唆されている26,33).しかしながら,R-Rasの上流のシグナル,すなわちどのような細胞外刺激でR-Rasが活性化されるのかということや,R-Rasがどのようにしてインテグリンを活性化するのかということはまったく不明であったため,まず,R-Rasがどのような細胞外刺激で活性されるのかを検討した.細胞接着因子受容体,インテグリンはコラーゲンやフィブロネクチンといったECMリガンドに結合し,活性化されることで細胞運動を促進する32)が,筆者らは,細胞が基質に接着しておらず脱着しているときには細胞内のR-Rasの活性が低いが,ひとたび細胞が接着し,ECMリガンドがインテグリンに結合すると,下流でR-Rasが活性化されるということを示した.また,このR-Rasの活性化がECMリガンドによって引き起こされるインテグリンの活性化や細胞運動に必要であるということを示した.そして,Sema4D/Plexin-B1によるR-Ras GAP活性はECMリガンドによるR-Rasの活性化を抑制し,結果としてECMによるインテグリンの活性化や細胞運動の促進を阻止することが明らかになった34).すなわち,R-RasはECMリガンドによって活性化され,活性化されたR-Rasがインテグリンの活性化を担うという,R-Ras活性とインテグリン活性の間の正のフィードバックループが存在する(図8)が,Sema4D/Plexin-B1によるR-Ras不活性化はECMリガンドによるR-Ras活性化を阻止することでそのループの1点を止め,細胞運動を阻止すると考えられる(図9).インテグリンを介した細胞接着は,細胞の最も基本的な機能の一つであり,Sema4D/Plexin-B1のR-Ras GAP活性は幅広い細胞種において基本的,普遍的な機能を担っているという示唆が得られた.
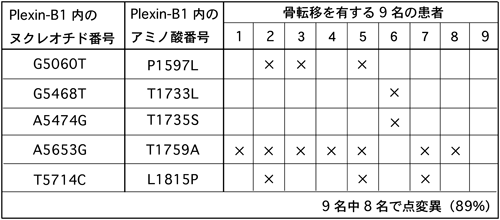
さらに,その後の筆者らと英国ロンドン大学のがん臨床チームとの共同研究で,このようなプレキシンのR-Ras GAP活性によるインテグリン活性制御のメカニズムは,正常細胞だけでなく,病態においても重要な役割を担っていることが明らかにされた.骨転移を有する前立腺がん患者由来の病理組織標本ではPlexin-B1の過剰発現が認められる.それらの遺伝子解析を行ったところ,高頻度にPlexin-B1の細胞内領域の点変異が認められ,それら点変異の場所は,Plexin-B1によるR-Ras GAP活性発揮に必要な機能ドメインであるC1領域およびC2領域の内部,あるいはRnd1の結合領域に位置していた(図10).それらの変異は,骨転移を有する患者9名中8名(89%)と高頻度であり,がん細胞の悪性度との連関を疑った.そこで,これらの変異型Plexin-B1を発現する細胞で細胞の運動能を検討したところ,いずれの変異型Plexin-B1もR-Ras GAP活性を発揮できず,細胞内のR-Ras活性が亢進されていることが明らかになった35).また,別の研究グループにより,R-Ras GAP活性の破綻による細胞内でのR-Ras活性亢進は,悪性黒色腫細胞の転移能獲得にも関わっていることも示された36).以上の知見から,プレキシンによるR-Ras GAP活性の情報伝達機構は,神経系に限らず幅広い細胞において,さらには,がん病態においても普遍的に駆動されるシステムであることが示唆された.
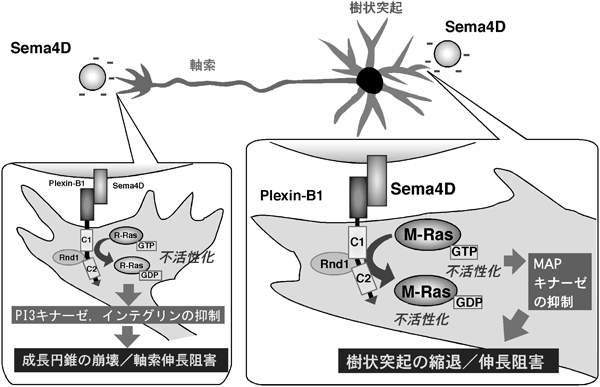
神経細胞に関しても,的確な回路網形成には軸索側と樹状突起側の双方の制御が必要である.的確な樹状突起伸展は,正確なシナプスおよび受容野の形成に重要であり,近年の研究により,神経成長因子や軸索ガイダンス因子などの細胞外因子によって,軸索側だけでなく,樹状突起側の形態調節が行われていることが明らかになってきている37).軸索ガイダンス因子の軸索に対する作用メカニズムの研究が進む一方,神経細胞がその周辺の因子を感知して自らの樹状突起の形や向きの変化を引き起こすメカニズムは明らかではなかった.筆者らのその後の研究で,内在性のR-Rasは海馬初代培養神経細胞において,軸索が伸長する時期に強く活性化され,また,その細胞内局在は軸索に偏在しており,R-Rasを欠失させた神経細胞では軸索の形成が著しく阻害されることが明らかになった38).このことから,R-Rasは軸索の伸長において決定的な役割を果たしていると考えられ,軸索においてプレキシンはこのR-Rasの活性を抑制することで反発性作用を発揮する.
Rasスーパーファミリーに属するR-Rasサブファミリーには,R-Ras(R-Ras1),TC21(R-Ras2),およびM-Ras(R-Ras3)という互いに構造上の相同性が高い3メンバーが属している39).しかしながら,高い相同性にも関わらず,R-RasとM-Rasはエフェクターの選択性や下流のシグナル伝達において違いがあることがわかっている.たとえば,R-Rasは下流の直接のエフェクターとしてPI3キナーゼ(phosphoinositide 3-kinase)を活性化することで細胞運動や神経突起伸長を正に制御するが,extracellular signal-regulated kinase(ERK)を活性化しない10,32).一方で,M-Rasは直接のエフェクターとしてB-Rafを活性化することでERK経路を活性化し,神経突起伸長を正に制御することが知られている40,41).R-Rasが組織や細胞種を問わず広範な発現を示すのに対し,M-Rasの発現は脳神経系,特に生後の大脳皮質や海馬での発現が高いことが知られていた40,42).そこでまず,Plexin-B1がR-Ras以外の他のR-RasファミリーメンバーにもGAP活性を発揮するかを検討したところ,R-Ras以外に,M-RasもプレキシンによるGAP活性の標的になることがわかった.大脳皮質および海馬の神経細胞において,個々のR-RasサブファミリーをRNA干渉法で発現抑制する実験により,軸索伸長はR-Rasが担うが,樹状突起伸長はR-RasではなくM-Rasが担っていることがわかった13).また,Sema4D刺激により,大脳皮質および海馬の神経細胞において,軸索だけでなく樹状突起においても突起の縮退という反発性応答が観察された.この反応は神経細胞内でのM-Ras活性の低下を伴っており,Plexin-B1によるGAP活性でM-Ras活性が低下し,下流でMAPキナーゼ経路の活性が抑制されることが,Sema4Dによって引き起こされる樹状突起の縮退に必要であることが明らかとなった13).以上から,神経細胞におけるガイダンス因子応答において,プレキシンによるGAP活性発揮のシステムが,軸索においてはR-Rasを標的とし,樹状突起においてはM-Rasを標的とすることで,軸索と樹状突起という二つの性質の異なる突起の反発性応答において,共通に駆動されるシステムであることが明らかになった(図11).
5. R-RasおよびM-Rasの下流のシグナル伝達経路
筆者らはプレキシンによるR-Ras GAP活性の下流で,PI3キナーゼの活性抑制やMAPキナーゼといったキナーゼ経路の活性抑制が引き起こされることが,軸索や樹状突起における反発作用に必要であることを明らかにした38,43).しかしながら,セマフォリンによる反発性応答の形態変化は,刺激後数分以内に起こる即時性の細胞骨格系の崩壊(セマフォリンの元の名であるコラプシンは“collapse=崩壊”から由来している)を伴う形態変化であることから,そのようなRas-キナーゼシグナル以外にも,より直接的に細胞骨格系を動かすシグナル経路が別にあるのではないかと,筆者らは考えた.微小管骨格制御に関しては,R-RasはPI3キナーゼシグナル経路を介して,チューブリンヘテロ二量体と結合して微小管の重合を促進する役割を持つ分子であるコラプシン反応介在タンパク質2(colapsin response mediator protein-2: CRMP-2)44)を活性化することで重合を制御していることが明らかになった45).一方で,R-Rasとアクチン骨格制御との直接的関係は,その後しばらく明らかではなかったが,筆者らの最近の研究で,R-Rasファミリーによるアクチン細胞骨格制御のエフェクターとしてラメリポディン(lamellipodin: Lpd)とアファディン(afadin)を同定した46,47).神経細胞において,これらはそれぞれR-Rasファミリーのエフェクターとして,前者は神経突起伸長を,後者は神経突起の分枝化を促進することが明らかになった.
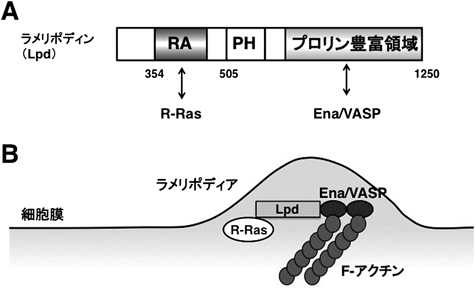
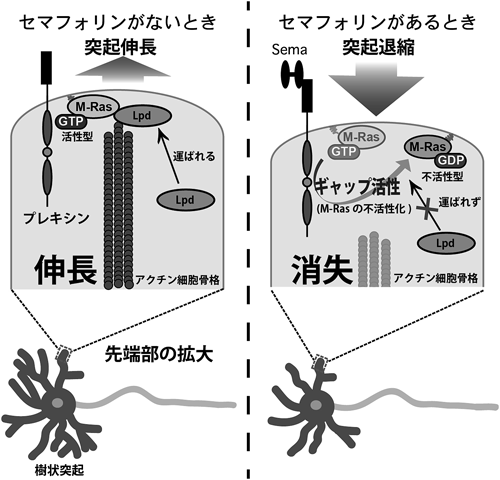
Lpdは抗アクチンキャッピングタンパク質であるEna/VASPの新奇結合タンパク質として同定された,アクチン重合を促進する約200 kDaのタンパク質である48).Lpdはアミノ末端側から,Ras結合ドメイン(RAドメイン),リン脂質結合ドメイン(PHドメイン),プロリン豊富領域を持ち,プロリン豊富領域内に六つ存在するFPPPモチーフを介して,Ena/VASP内のEVH1ドメイン(プロリン残基を多く含む配列を認識し結合するドメイン)と結合することで,Ena/VASPの細胞内局在を制御し,アクチン骨格を制御している(図12).Ena/VASPは葉状仮足(ラメリポディア)や糸状仮足(フィロポディア)の先端で,アクチンキャッピングタンパク質と拮抗することにより,アクチン重合を正に制御することが知られている49).また,Ena/VASPを欠損させた神経細胞は,突起を伸展することができないことから,Ena/VASPによる抗アクチンキャッピング作用は神経突起の形成および伸長に必須であると考えられている50).線虫におけるLpdに相当する分子であるMig-10は,神経細胞において移動,極性形成,軸索ガイダンス因子のネトリンやslitによる軸索ガイダンス,さらにはシナプス小胞の形成という,神経機能発揮におけるさまざまな局面で重要な役割を果たしている51–54).Lpdは単独では細胞質に豊富に存在するタンパク質であるが,細胞膜に局在する活性化型のR-RasやM-Rasとの結合によって膜移行される.この膜移行のステップが神経突起先端でのアクチン重合の促進に必須であるが,反発性ガイダンス因子Sema4Dは,Plexin-B1によるR-Ras GAP活性によりR-Rasファミリーを不活性化型にすることで,この膜移行を阻害する.このことにより,神経突起先端でアクチン線維の消失が生じ,神経線維の退縮や反発応答が起こることが明らかになった46)(図13).
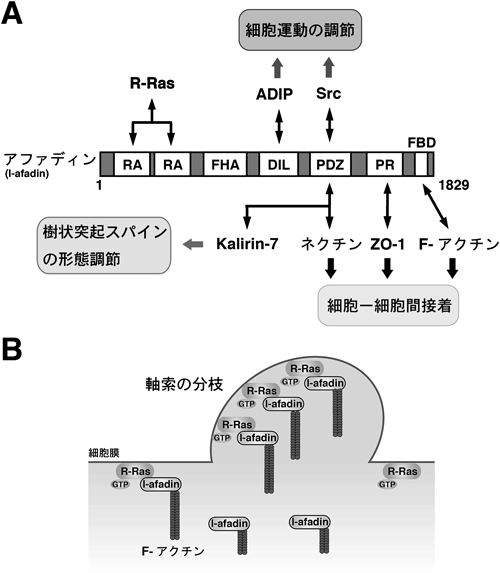
また,大脳皮質神経細胞においてSema3Aは軸索の分枝形成を抑制することがわかっているが55),そのメカニズムは明らかではなかった.アファディン(別名AF-6,canoe,またはMLLT4)はRAドメインやPDZドメインを持つ,アダプタータンパク質である56)(図14A).元来,アファディンは非神経細胞の細胞—細胞間接着部位においてF-アクチン(filamentous-actin)と結合する分子として同定された57).上皮細胞においては,アファディンは細胞間接着分子であるネクチン(nectin)と結合し,ネクチンとアクチン骨格のリンカーとして働くことで,細胞—細胞間接着の形成に関与していることがわかっている58,59).ネクチン以外にも,アファディンは細胞—細胞間接着の形成および安定化に働くさまざまな分子と結合することが知られている60–62).細胞—細胞間接着制御以外にも,最近の研究により,アファディンは乳がん細胞や線維芽細胞において細胞運動を制御していることもわかっている63,64).神経細胞では,アファディンは樹状突起スパインの形態を制御していることが細胞レベル,さらには個体レベルの実験系で明らかとなっている65,66).一方で,アファディンは軸索にも局在していることがわかっているが67),軸索の形態制御における機能は明らかではなかった.筆者らの研究で,軸索成長期の神経細胞においてR-Rasとアファディンは内在的に結合し,R-Rasは活性型特異的にアファディンのRAドメインと結合することがわかった.大脳皮質初代培養神経細胞の培養後4日目までの軸索成長期において,アファディンのRNA干渉法によるノックダウンおよび過剰発現を行ったところ,ノックダウンにより軸索分枝が減少し,過剰発現により軸索分枝が増加した.また,活性型R-Rasによって顕著な軸索分枝形成の増加が引き起こされるが,その増加分はアファディンのノックダウンによって阻止された.以上から,活性化されたR-Rasはアファディンを介して軸索分枝形成を行うことが明らかになった47).アファディンもLpd同様に,単独では細胞質に存在するタンパク質であるが,活性型R-Rasのアファディンへの結合は,アファディンを細胞質から細胞膜へと移行させた.また,細胞膜移行シグナルを付加したアファディンの変異体を過剰発現させた神経細胞では軸索の分枝化が促進されたことから,アファディンは細胞膜移行されることにより,軸索分枝形成能を発揮することがわかった68).また,大脳皮質初代培養神経細胞の内在性のアファディンは軸索先頂部や軸索幹部の分枝形成部位の成長円錐に,F-アクチンとともに集積しており,アファディンによって引き起こされる軸索分枝形成はアクチン重合阻害剤により阻止された.以上の結果から,活性型R-Rasがアファディンを細胞膜へ運び,運ばれたアファディンがその場でアクチン骨格の再構築を促すことで軸索分枝形成を行うという,Rasタンパク質を介した軸索分枝形成の新しい分子メカニズムが明らかになった(図14B).
R-Rasファミリーの下流分子としてのLpdやアファディンに関するこれらの知見は,従来,MAPキナーゼやPI3キナーゼなど,キナーゼシグナルの活性化因子として周知され,がん原遺伝子として名高いRasタンパク質が,細胞骨格制御タンパク質と直接結合しその活性を制御することで細胞形態を調節しているという,新たな知見を与えるものである.
これまでの報告により,さまざまな細胞外因子や神経細胞の活動性が,神経細胞の形態に影響を与えることがわかっている69–71).たとば,ネトリン-1およびfibroblast growth factor-2(FGF-2)は軸索の分枝形成を促進し,反発性軸索ガイダンス因子のSema3Aは逆に分枝形成を抑制することが大脳皮質神経細胞を用いた研究によって明らかになっている72).また,別の反発性軸索ガイダンス因子のエフリンも軸索の分枝形成の抑制や成長円錐の崩壊といった,神経軸索における反発性応答や細胞の基質からの脱着に関与している71).筆者らの研究で,神経伸長や基質との接着を正に制御するECMリガンドはその受容体であるβ1インテグリンに結合して細胞内でR-Rasを活性化し,一方で,神経伸長を負に制御するセマフォリンはその受容体であるプレキシンのR-Ras GAP活性により,R-Rasを直接不活性化することで反発性応答を発揮することが明らかとなった11,28,29,34).別の反発性ガイダンス因子のエフリンに関しても,エフリンの受容体であるEph受容体にR-Rasに対するGAPが結合し,リガンド依存的に活性化されることにより細胞内のR-Ras活性を低下させることで軸索の退縮や細胞の脱着を引き起こすことも報告されている73).また,神経細胞において,誘引性ガイダンス因子の一つであるネトリン-1はアデノシンA2b受容体を介して細胞内でのcAMP濃度を上昇させることがわかっており74),非神経系の培養細胞を用いた研究で,cAMP濃度の上昇はR-Rasに対するGEFの一つ,cAMP-GEF(Epac)の活性化を引き起こし,細胞内のR-Ras活性上昇を引き起こすことがわかっている75).また,筆者らのごく最近の研究により,大脳皮質神経細胞における細胞内cAMP濃度の上昇は,神経細胞内在性のR-Rasの活性化を引き起こすこと,また,この活性化によりアファディンの細胞膜移行が引き起こされ,軸索の分枝化が引き起こされることが明らかになった68).
以上の知見から,神経伸長や分枝化,細胞の接着や運動に正負に働くさまざまなガイダンス因子の刺激が,細胞内においてR-Rasの活性に統合され,R-Rasがいわばシグナル伝達のハブとして働き,下流でキナーゼ系,骨格制御系を含めたさまざまなエフェクターの活性を並行に制御することで,最終的な細胞形態が決定されているものと示唆される.これまでにも,個々のガイダンス因子を標的とした神経再生やがん転移抑制のための創薬研究は行われてきているが,筆者らは,ガイダンスシグナルの集約地点であるR-Ras活性を制御できるシステムができれば,複数のガイダンス因子シグナルを一手にコントロールすることが可能になり,より効率的な神経再生,がん転移抑制を可能とする創薬が可能になると考えている.今後も,そのシステムの構築を目指して研究を続けていきたい.
謝辞Acknowledgments
本稿で紹介した研究は,京都大学大学院生命科学研究科生体システム学分野において行われたものであり,研究室を主宰される根岸学先生に心から感謝いたします.転移性前立腺がん患者におけるPlexin-B1の点変異解析に関する研究は,英国ロンドン大学のMagali Williamson博士との共同研究でなされたもので,国際共同研究の貴重な機会に心から感謝します.また,戦略的創造研究推進事業さきがけ「脳神経回路の形成・動作と制御領域」の総括の村上富士夫先生(大阪大学)や,領域アドバイザーの先生がたの真摯なご助言,さらに領域研究者の戸島拓郎先生(理研BSI)や松田孝彦先生(京都大学)の日常の討論,技術的サポート,さらにはたゆまぬ激励にも,深く感謝いたします.
最後に,これら一連の研究は,生体システム学分野に所属していた大学院生とともに実験し,一喜一憂,苦楽をともにし,一つずつ積み重ねられてきた成果です.ここに,あらためて,関係者の皆様に深く御礼を申し上げます.
引用文献References
1) Ramón y Cajal, S. (1892) Cellule, 9, 119–258.
2) Carmeliet, P. & Tessier-Lavigne, M. (2005) Nature, 436, 193–200.
3) Kruger, R.P., Aurandt, J., & Guan, K.L. (2005) Nat. Rev. Mol. Cell Biol., 6, 789–800.
4) Takayanagi, H. (2007) Nat. Rev. Immunol., 7, 292–304.
5) Kumanogoh, A. & Kikutani, H. (2013) Nat. Rev. Immunol., 13, 802–814.
6) Corà, D., Astanina, E., Giraudo, E., & Bussolino, F. (2014) Trends Mol. Med., 20, 589–598.
7) Eissler, N. & Rolny, C. (2013) Exp. Cell Res., 319, 1635–1643.
8) Ivins, J.K., Yurchenco, P.D., & Lander, A.D. (2000) J. Neurosci., 20, 6551–6560.
9) Keely, P.J., Rusyn, E.V., Cox, A.D., & Parise, L.V. (1999) J. Cell Biol., 145, 1077–1088.
10) Kinbara, K., Goldfinger, L.E., Hansen, M., Chou, F.L., & Ginsberg, M.H. (2003) Nat. Rev. Cell Biol., 4, 767–776.
11) Oinuma, I., Ishikawa, Y., Katoh, H., & Negishi, M. (2004) Science, 305, 862–865.
12) Luo, Y., Raible, D., & Raper, J.A. (1993) Cell, 75, 1389–1399.
13) Tamagnone, L., Artigiani, S., Chen, H., He, Z., Ming, G.I., Song, H., Chedotal, A., Winberg, M.L., Goodman, C.S., Poo, M., Tessier-Lavigne, M., & Comoglio, P.M. (1999) Cell, 99, 71–80.
14) Swiercz, J.M., Kuner, R., Behrens, J., & Offermanns, S. (2002) Neuron, 35, 51–63.
15) Hirotani, M., Ohoka, Y., Yamamoto, T., Nirasawa, H., Furuyama, T., Kogo, M., Matsuya, T., & Inagaki, S. (2002) Biochem. Biophys. Res. Commun., 297, 32–37.
16) Aurandt, J., Vikis, H.G., Gutkind, J.S., Ahn, N., & Guan, K.L. (2002) Proc. Natl. Acad. Sci. USA, 99, 12085–12090.
17) Perrot, V., Vazquez-Prado, J., & Gutkind, J.S. (2002) J. Biol. Chem., 277, 43115–43120.
18) Oinuma, I., Katoh, H., Harada, A., & Negishi, M. (2003) J. Biol. Chem., 278, 25671–25677.
19) Nakamura, F., Kalb, R.G., & Strittmatter, S.M. (2000) J. Neurobiol., 44, 219–229.
20) Rhom, B., Rahim, B., Kleiber, B., Hovatta, I., & Püschel, A.W. (2000) FEBS Lett., 486, 68–72.
21) Raper, J.A. (2000) Curr. Opin. Neurobiol., 10, 88–94.
22) Trusolino, L. & Comoglio, P.M. (2002) Nat. Rev. Cancer, 2, 289–300.
23) Goshima, Y., Ito, T., Sasaki, Y., & Nakamura, F. (2002) J. Clin. Invest., 109, 993–998.
24) Hu, H., Marton, T.F., & Goodman, C.S. (2001) Neuron, 32, 53–62.
25) Scheffzek, K., Ahmandian, M., & Wittinghofer, A. (1998) Trends Biochem. Sci., 23, 257–262.
26) Zhang, Z., Vuori, K., Wang, H., Reed, J.C., & Ruoslahti, E. (1996) Cell, 85, 61–69.
27) Zanata, S.M., Hovatta, I., Rhom, B., & Püschel, A.W. (2002) J. Neurosci., 22, 471–477.
28) Toyofuku, T., Yoshida, J., Sugimoto, T., Zhang, H., Kumanogoh, A., Hori, M., & Kikutani, H. (2005) Nat. Neurosci., 8, 1712–1719.
29) Uesugi, K., Oinuma, I., Katoh, H., & Negishi, M. (2009) J. Biol. Chem., 284, 6743–6751.
30) Barberis, D., Artigiani, S., Casazza, A., Corso, S., Giordano, C., Love, C.A., Jones, E.Y., Comoglio, P.M., & Tamagnone, L. (2004) FASEB J., 18, 592–594.
31) Walzer, T., Galibert, L., Comeau, M.R., & De Smedt, T. (2005) J. Immunol., 174, 51–59.
32) Hood, J.D. & Cheresh, D.A. (2002) Nat. Rev. Cancer, 2, 91–100.
33) Sethi, T., Ginsberg, M.H., Downward, J., & Hughes, P.E. (1999) Mol. Biol. Cell, 10, 1799–1809.
34) Oinuma, I., Katoh, H., & Negishi, M. (2006) J. Cell Biol., 173, 601–613.
35) Wong, O.G., Nitkunan, T., Oinuma, I., Zhou, C., Blanc, V., Brown, R.S., Bott, S.R., Nariculam, J., Box, G., Munson, P., Constantinou, J., Feneley, M.R., Klocker, H., Eccles, S.A., Negishi, M., Freeman, A., Masters, J.R., & Williamson, M. (2007) Proc. Natl. Acad. Sci. USA, 104, 19040–19045.
36) Chen, Y., Soong, J., Mohanty, S., Xu, L., & Scott, G. (2013) Oncogene, 32, 4941–4949.
37) Parrish, J.Z., Emoto, K., Kim, M.D., & Jan, Y.N. (2007) Annu. Rev. Neurosci., 30, 399–423.
38) Oinuma, I., Katoh, H., & Negishi, M. (2007) J. Biol. Chem., 282, 303–318.
39) Matsumoto, K., Asano, T., & Endo, T. (1997) Oncogene, 15, 2409–2417.
40) Kimmelman, A.C., Nuñez Rodriguez, N., & Chan, A.M. (2002) Mol. Cell. Biol., 22, 5946–5961.
41) Sun, P., Watanabe, H., Takano, K., Yokoyama, T., Fujisawa, J., & Endo, T. (2006) Genes Cells, 11, 1097–1113.
42) Rodriguez-Viciana, P., Warne, P.H., Dhand, R., Vanhaesebroeck, B., Gout, I., Fry, M.J., Waterfield, M.D., & Downward, J. (1994) Nature, 370, 527–532.
43) Saito, Y., Oinuma, I., Fujimoto, S., & Negishi, M. (2009) EMBO Rep., 10, 614–621.
44) Fukata, Y., Itoh, T.J., Kimura, T., Ménager, C., Nishimura, T., Shiromizu, T., Watanabe, H., Inagaki, N., Iwamatsu, A., Hotani, H., & Kaibuchi, K. (2002) Nat. Cell Biol., 4, 583–591.
45) Ito, Y., Oinuma, I., Katoh, H., Kaibuchi, K., & Negishi, M. (2006) EMBO Rep., 7, 704–709.
46) Tasaka, G., Negishi, M., & Oinuma, I. (2012) J. Neurosci., 32, 8293–8305.
47) Iwasawa, N., Negishi, M., & Oinuma, I. (2012) Mol. Biol. Cell, 23, 2793–2804.
48) Krause, M., Leslie, J.D., Stewart, M., Lafuente, E.M., Valderrama, F., Jagannathan, R., Strasser, G.A., Rubinson, D.A., Liu, H., Way, M., Yaffe, M.B., Boussiotis, V.A., & Gertler, F.B. (2004) Dev. Cell, 7, 571–583.
49) Krause, M., Dent, E.W., Bear, J.E., Loureiro, J.J., & Gertler, F.B. (2003) Annu. Rev. Cell Dev. Biol., 19, 541–564.
50) Kwiatkowski, A.V., Rubinson, D.A., Dent, E.W., Edward van Veen, J., Leslie, J.D., Zhang, J., Mebane, L.M., Philippar, U., Pinheiro, E.M., Burds, A.A., Bronson, R.T., Mori, S., Fässler, R., & Gertler, F.B. (2007) Neuron, 56, 441–455.
51) Adler, C.E., Fetter, R.D., & Bargmann, C.I. (2006) Nat. Neurosci., 9, 511–518.
52) Chang, C., Adler, C.E., Krause, M., Clark, S.G., Gertler, F.B., Tessier-Lavigne, M., & Bargmann, C.I. (2006) Curr. Biol., 16, 854–862.
53) Quinn, C.C., Pfeil, D.S., Chen, E., Stovall, E.L., Harden, M.V., Gavin, M.K., Forrester, W.C., Ryder, E.F., Soto, M.C., & Wadsworth, W.G. (2006) Curr. Biol., 16, 845–853.
54) Stavoe, A.K. & Colón-Ramos, D.A. (2012) J. Cell Biol., 197, 75–88.
55) Dent, E.W., Barnes, A.M., Tang, F., & Kalil, K. (2004) J. Neurosci., 24, 3002–3012.
56) Takai, Y., Ikeda, W., Ogita, H., & Rikitake, Y. (2008) Annu. Rev. Cell Dev. Biol., 24, 309–342.
57) Mandai, K., Nakanishi, H., Satoh, A., Obaishi, H., Wada, M., Nishioka, H., Itoh, M., Mizoguchi, A., Aoki, T., Fujimoto, T., Matsuda, Y., Tsukita, S., & Takai, Y. (1997) J. Cell Biol., 139, 517–528.
58) Takahashi, K., Nakanishi, H., Miyahara, M., Mandai, K., Satoh, K., Satoh, A., Nishioka, H., Aoki, J., Nomoto, A., Mizoguchi, A., & Takai, Y. (1999) J. Cell Biol., 145, 539–549.
59) Kurita, S., Ogita, H., & Takai, Y. (2011) J. Biol. Chem., 286, 36297–36303.
60) Tachibana, K., Nakanishi, H., Mandai, K., Ozaki, K., Ikeda, W., Yamamoto, Y., Nagafuchi, A., Tsukita, S., & Takai, Y. (2000) J. Cell Biol., 150, 1161–1176.
61) Pokutta, S., Drees, F., Takai, Y., Nelson, W.J., & Weis, W.I. (2002) J. Biol. Chem., 277, 18868–18874.
62) Asada, M., Irie, K., Morimoto, K., Yamada, A., Ikeda, W., Takeuchi, M., & Takai, Y. (2003) J. Biol. Chem., 278, 4103–4111.
63) Miyata, M., Ogita, H., Komura, H., Nakata, S., Okamoto, R., Ozaki, M., Majima, T., Matsuzawa, N., Kawano, S., Minami, A., Waseda, M., Fujita, N., Mizutani, K., Rikitake, Y., & Takai, Y. (2009) J. Cell Sci., 122, 4319–4329.
64) Fournier, G., Cabaud, O., Josselin, E., Chaix, A., Adélaïde, J., Isnardon, D., Restouin, A., Castellano, R., Dubreuil, P., Chaffanet, M., Birnbaum, D., & Lopez, M. (2011) Oncogene, 30, 3862–3874.
65) Xie, Z., Huganir, R.L., & Penzes, P. (2005) Neuron, 48, 605–618.
66) Beaudoin, G.M. 3rd, Schofield, C.M., Nuwal, T., Zang, K., Ullian, E.M., Huang, B., & Reichardt, L.F. (2012) J. Neurosci., 32, 99–110.
67) Lim, S.T., Lim, K.C., Giuliano, R.E., & Federoff, H.J. (2008) J. Comp. Neurol., 507, 1228–1244.
68) Umeda, K., Iwasawa, N., Negishi, M., & Oinuma, I. (2015) Mol. Biol. Cell, in press.
69) Dent, E.W. & Kalil, K. (2001) J. Neurosci., 21, 9757–9769.
70) Yamada, A., Uesaka, N., Hayano, Y., Tabata, T., Kano, M., & Yamamoto, N. (2010) Proc. Natl. Acad. Sci. USA, 107, 7562–7567.
71) Bilimoria, P.M. & Bonni, A. (2013) Neuroscientist, 19, 16–24.
72) Dent, E.W., Barnes, A.M., Tang, F., & Kalil, K. (2004) J. Neurosci., 24, 3002–3012.
73) Dail, M., Richter, M., Godement, P., & Pasquale, E.B. (2006) J. Cell Sci., 119, 1244–1254.
74) Corset, V., Nguyen-Ba-Charvet, K.T., Forcet, C., Moyse, E., Chédotal, A., & Mehlen, P. (2000) Nature, 407, 747–750.
75) López De Jesús, M., Stope, M.B., Oude Weernink, P.A., Mahlke, Y., Börgermann, C., Ananaba, V.N., Rimmbach, C., Rosskopf, D., Michel, M.C., Jakobs, K.H., & Schmidt, M. (2006) J. Biol. Chem., 281, 21837–21847.
著者紹介Author Profile
 生沼 泉(おいぬま いずみ)
生沼 泉(おいぬま いずみ)京都大学助教(大学院生命科学研究科).博士(生命科学).
略歴1978年青森県に生る.2002年京都大学薬学部総合薬学科卒業.04年同大学大学院生命科学研究科修士課程修了.07年同博士後期課程修了(DC1).07年より現職.11年より同大学薬学研究科助教(兼担),JSTさきがけ研究者(兼任).
研究テーマと抱負ガイダンス因子シグナルの普遍的動作原理の解明を目指している.神経繊維再生やがん治療への応用の基盤研究として,低分子量Gタンパク質を基軸に,派手さはなくとも骨太でしっかりとした研究を積み重ねていきたい.
趣味愛犬(シーリハムテリア),音楽鑑賞・演奏(クラシック),ラテン語(ローマ哲学).