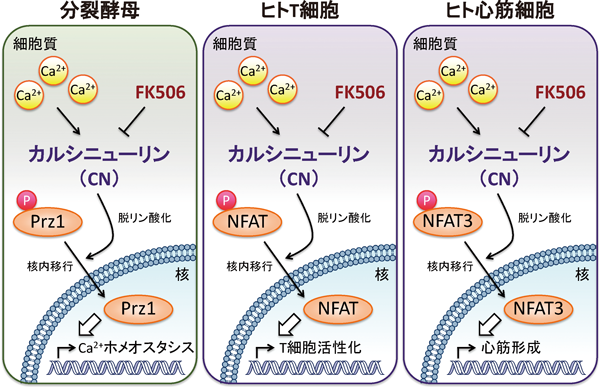
すべての真核生物に高度に保存されているカルシニューリンは,Ca2+/カルモジュリン依存性に活性化されるSer/Thrホスファターゼであり,カルシウム(Ca2+)シグナル伝達のキープレーヤーである1).1991年に,免疫抑制薬タクロリムス(FK506)/シクロスポリンAが,それぞれのイムノフィリンと結合し,カルシニューリンの酵素活性を阻害することによりその免疫抑制効果を発揮することが発見されて以来,カルシニューリンは,臓器移植に不可欠な免疫抑制薬の標的として,またCa2+シグナルを仲介する重要なシグナル伝達分子として脚光を集めてきた2,3).最も詳細にカルシニューリンシグナルが解析されているT細胞において,カルシニューリンは転写因子NFAT(nuclear factor of activated T cells)を脱リン酸化し,NFATの核内移行を引き起こすことでIL-2などのサイトカインの発現を誘導することが明らかとなった4).
このような,「Ca2+–カルシニューリン–転写因子–遺伝子発現制御」という“シグナル伝達カセット”,ならびに“カルシニューリンによる転写因子の核内移行制御”というメカニズムは酵母から高等生物まで進化的に高度に保存されている(図1).特に,心筋細胞において,FK506投与により心筋肥大が抑制されるという報告がなされて以来,カルシニューリンシグナルの制御は心肥大治療薬という新たな疾患治療のターゲットとしても注目されるようになった5,6).また,カルシニューリンの特異的阻害薬であるFK506は,臓器移植に伴う拒絶反応の予防に不可欠であるが,近年アトピー性皮膚炎を始めとする湿疹治療においても著効を発揮するなど,その医療的用途は拡大している.
一方,免疫抑制薬であるFK506を投与されたときに,腎障害,高脂血症,多毛,高血圧などが副作用として高頻度で生じることが報告されているが,これらも生体内でカルシニューリンの酵素活性が抑制された結果生じると考えられることから,カルシニューリンはさまざまな分子との機能的相互作用を介して,その生理機能を発揮していると推測される.しかしながらこのようなカルシニューリンの生理的な役割,あるいはカルシニューリンの作用経路を分子レベルで明らかにすることは容易ではない.
我々は,遺伝学的手法が駆使でき,高等生物にきわめて近い細胞内シグナル伝達経路を有する分裂酵母をモデル生物としてカルシニューリン研究を行ってきた7).特に,カルシニューリンとMAPK経路の機能的相互作用の発見に立脚した遺伝学的戦略を展開することにより,カルシニューリンとクロストークするさまざまなシグナル伝達経路を解明してきた8–12).さらに,分裂酵母においてもカルシニューリンがFK506の標的分子であることに着目した「ケミカルゲノミクス」を駆使して免疫抑制薬感受性遺伝子群を同定し,カルシニューリンの新たな細胞機能を次々と明らかにした13–21).
一方,前述のとおり,カルシニューリンによる標的転写因子の細胞内局在・遺伝子発現の制御機構に関しては,さまざまな細胞や生物種で詳細な研究がなされているにも関わらず,カルシニューリン自身の細胞内局在がどのような機構で調節されているのかということはほとんど解明されていなかった.我々は,ストレス刺激に応答したカルシニューリンの細胞内局在を解析した結果,カルシニューリンがストレス依存的に劇的な局在の変化を示し,「RNA顆粒」と呼ばれる細胞質内の構造体に移行することを見いだした22).RNA顆粒は,RNAとその代謝などに関わる分子を中心に形成されることから,当初「RNAを介する遺伝子発現制御」という役割のみが注目を集めていた.しかしながら,近年TOR(target of Rapamycin)をはじめとしたさまざまなシグナル分子がRNA顆粒にリクルートされること,さらに,がんやアルツハイマー病,パーキンソン病,ウイルス感染などの疾患や病態との密接な関わりが明らかになったことから,RNA顆粒は「シグナル伝達の拠点」として,また「疾患治療の標的」としても新たな注目を集めている23).本稿では,我々の研究から浮かび上がってきたカルシニューリンの新たな空間的制御メカニズムと,RNA顆粒形成の疾患治療への応用の可能性について紹介する.
2. カルシニューリンとMAPキナーゼ経路の拮抗的なクロストークを利用したMAPキナーゼシグナル伝達経路の制御因子群の同定
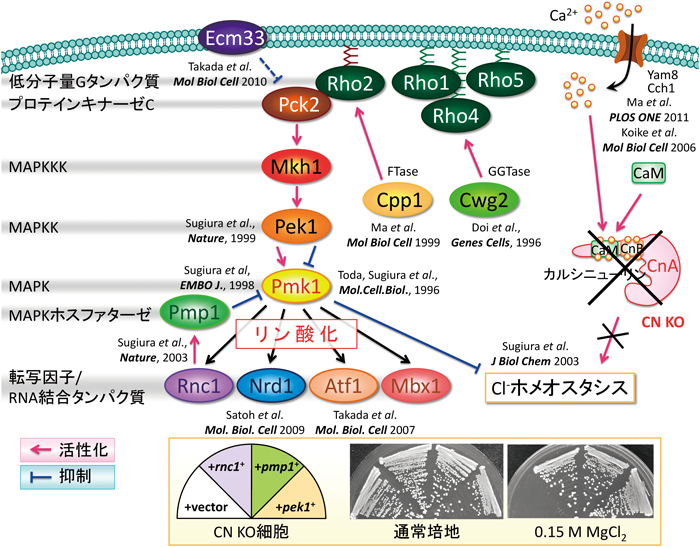
カルシニューリン(図2,CN)は触媒サブユニット(CnA)とCa2+結合制御サブユニット(CnB)から構成されるヘテロ二量体であり,細胞内Ca2+濃度が上昇するとCa2+結合タンパク質であるカルモジュリン(図2,CaM)がカルシニューリンに結合することで活性化される(図2).免疫抑制薬であるFK506はFKBP12,シクロスポリンAはシクロフィリン(CyP)というそれぞれのイムノフィリンと複合体を作り,カルシニューリンの活性を阻害する(図2).
分裂酵母では,触媒サブユニットであるPpb1も制御サブユニットであるCnb1も1種類の遺伝子にコードされており,どちらの遺伝子をノックアウトしても同じ表現型を示す.また,Ppb1とCnb1を同時にノックアウトしても単独ノックアウト細胞と表現型は同じであり,触媒サブユニットを過剰発現しても制御サブユニットがなければ機能を発揮できない24).我々は,遺伝学とケミカルゲノミクスを駆使でき,かつ最もシンプルなカルシニューリン研究のモデル生物として分裂酵母を用いて研究を行ってきた.特にカルシニューリンノックアウト,あるいはFK506処理によるカルシニューリン酵素活性阻害に伴う表現型である「Cl−感受性」を利用することにより,高等生物のERK MAPK(extracellular signal-regulated MAP kinase)のホモログであるPmk1 MAPK経路の制御因子群を網羅的に同定する手法を確立した(詳細は他の我々の総説25)あるいはHenry Stewart Talks Onlineを参照されたい).
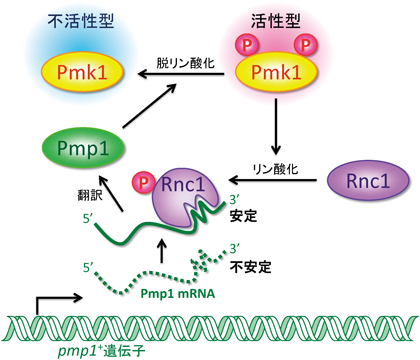
このようなカルシニューリンとMAPKの拮抗的な機能的関係を利用した遺伝学的スクリーニングの結果,Pmk1 MAPK経路の抑制制御因子群として,MAPKを脱リン酸化し,負に制御する二重特異性MAPKホスファターゼPmp1,プロテインホスファターゼ2C Ptc1,Ptc3,さらにPmp1 mRNAを安定化するKH型RNA結合タンパク質Rnc1あるいは細胞表面膜タンパク質であるEcm33などを同定した(図3)8,10,26,27).
また,同様の遺伝学的スクリーニングによって,MAPKの活性化因子であるMAP kinase kinase Pek1,MAP kinase kinase kinase Mkh1,ファルネシルトランスフェラーゼCpp1,ゲラニルゲラニルトランスフェラーゼCwg2,低分子量Gタンパク質Rho2などを同定した(図3)9,11,12).
これらの制御因子は種を超えて高度に保存されていると同時に,MAPKシグナルの活性制御に関わり,ヒトにおける発がんのメカニズムに直結することからも,きわめて魅力的な創薬の標的分子でもある.実際に我々はこの表現型を活用した創薬探索研究を行い,抗がん薬シーズの同定に成功している25,28).
3. ケミカルゲノミクスを利用した合成致死スクリーニングとmRNA結合タンパク質Nrd1の同定
我々の用いたもう一つの遺伝学的戦略は,“FK506を用いたケミカルゲノミクス”である.このスクリーニングを通して,mRNA結合タンパク質Nrd1を同定したが,結果的にNrd1はPmk1 MAPKの標的因子であると同時に,後述するRNA顆粒の重要な制御因子であることが判明した29,30).
我々の行ったケミカルゲノミクススクリーニングは,FK506が分裂酵母のカルシニューリンを臨床用量で阻害することに着目した「FK506感受性原因遺伝子群の同定」である.このスクリーニングのロジックを示す.分裂酵母の正常細胞に対してFK506を添加しても(あるいはカルシニューリン遺伝子をノックアウトしても),細胞増殖に影響はない.しかしながら,もしもカルシニューリンと,増殖に必須な生命現象をシェアするような遺伝子Xに突然変異が入った変異体が存在した場合には,この変異体XはFK506に感受性を示すと予想される.すなわち,変異体Xの増殖にはカルシニューリンの正常な機能が不可欠である.これは,「合成致死(synthetic lethal)」と呼ばれ,遺伝子XとY(この場合カルシニューリン)が生命現象に必須な機能をシェアしていることを示唆する遺伝学的な解析手法である.したがって,FK506感受性遺伝子群を網羅的に同定することにより,カルシニューリンが関わる新たな生命現象やカルシニューリンと機能的に関連する遺伝子群を明らかにできる.我々は,FK506感受性原因遺伝子として,表1に示すような遺伝子群を同定し,細胞質分裂,細胞内輸送,イノシトールリン脂質代謝といった生命機能の制御にカルシニューリンが密接に関わることを明らかにしてきた.その中の一種,its7+/cdc4+遺伝子は,ミオシン軽鎖をコードしており,細胞質分裂に必須の役割をする.我々は,cdc4変異体の示す細胞質分裂異常を回復できる遺伝子として,mRNA結合タンパク質Nrd1を同定し,Nrd1がCdc4 mRNAと結合し,安定化することにより,細胞質分裂を制御することを明らかにした29).興味深いことに,Nrd1はPmk1 MAPKによってリン酸化されることにより,RNA結合活性が負に制御される.すなわち,Nrd1はMAPKの標的としてCdc4 mRNAを始めとする細胞質分裂に関わる遺伝子発現の鍵を握る29).このような「MAPK依存的なmRNA結合タンパク質の活性制御」というメカニズムは,前述のMAPKホスファターゼmRNAを標的とするRnc1に加え(図4),高等生物においても数多くの報告がある31).したがって「MAPKを介するシグナル伝達と遺伝子発現の担い手」としてmRNA結合タンパク質は,普遍的に重要な役割を果たしていることが明らかになった.
表1 FK506感受性原因遺伝子群とその細胞内機能| 遺伝子名 | 遺伝子産物 | 細胞内機能 | 参考文献 |
|---|
| its1+/apl2+ | β-アダプチン | 細胞内小胞輸送 | Ma, Sugiura et al., Genes Cells 2009 |
| its3+ | PI4P5キナーゼ | 脂質代謝,細胞内小胞輸送 | Zhang, Sugiura et al., J. Biol. Chem. 2000 |
| its4+/sip1+ | AP-1付属タンパク質 | 細胞内小胞輸送 | Yu, Sugiura et al., PLoS ONE 2012 |
| its5+/ypt3+ | Rabファミリー低分子量Gタンパク質 | 細胞内小胞輸送 | Cheng, Sugiura et al., Mol. Biol. Cell 2002 |
| its6+/ryh1+ | Rabファミリー低分子量Gタンパク質 | 細胞内小胞輸送 | He, Sugiura et al., Genes Cells 2006 |
| its7+/cdc4+ | ミオシンⅡ必須軽鎖 | 細胞質分裂,細胞骨格 | Fujita, Sugiura et al., Genetics 2002Satoh, Sugiura et al., Mol. Biol. Cell 2009 |
| its8+ | ヒトPig-nホモログ | GPIアンカー合成 | Yada, Sugiura et al., J. Biol. Chem. 2001 |
| its9+ | クラスリン重鎖 | 細胞内小胞輸送 | |
| its10+/cdc7+ | タンパク質リン酸化酵素 | 細胞質分裂 | Lu, Sugiura et al., Genes Cells 2002 |
| its11+/gdi1+ | Rab-GDP解離抑制因子(GDI) | 細胞内小胞輸送 | Ma, Sugiura et al., Genetics 2006 |
| vas1+/vps45+ | Vps45ホモログ | 細胞内小胞輸送 | Miyatake, Sugiura et al., Genetics 2007 |
| vas2+/aps1+ | σ-アダプチン | 細胞内小胞輸送 | Ma, Sugiura et al., Genes Cells 2009 |
| cis1+/amp1+ | μ-アダプチン | 細胞内小胞輸送 | Kita, Sugiura et al., Mol. Biol. Cell 2004 |
| cis2+/myp2+ | ミオシンⅡ重鎖 | 細胞質分裂,細胞骨格 | Fujita, Sugiura et al., Genetics 2002 |
| cis3+/imp2+ | PSTPIPホモログ | 細胞骨格,細胞内小胞輸送 | Kita, Sugiura et al., Biochem. Biophys. Res. Commun. 2014 |
このようにして同定されたNrd1はRRM(RNA recognition motif)型のRNA結合タンパク質をコードしており,高等生物において相同性の高いRNA結合タンパク質として,TIA-1(T-cell internal antigen-1)/TIAR(TIA-1-related protein)が存在する.Nrd1とTIA-1/TIARは,アミノ酸レベルで相同性を示すのみならず,TIA-1/TIARが認識するRNAコンセンサス配列(UCUU)をNrd1も同様に認識することから,Nrd1がTIA-1の機能的オルソログではないかと推測した29).興味深いことに,TIA-1/TIARはRNA顆粒の一種であるストレス顆粒(stress granules: SG)の代表的なマーカータンパク質であり,高等生物におけるSG形成の中心的な役割を果たすことが知られている.
RNA顆粒は,さまざまなストレス刺激に応答してダイナミックに形成される細胞内構造体であり,RNAの輸送や分解,翻訳などのRNA代謝を時空間的にリプログラミングする.RNA顆粒にはさまざまな種類が存在するが,代表的なものとして,ストレス時のRNA代謝制御に関わるSGと,主としてmRNA分解や翻訳抑制に関わるP-body(processing body)があり,その構成因子や制御機構は種を超えて高度に保存されている32).RNA顆粒は膜を持たない凝集体であり,その構成要素としてmRNA,RNA結合タンパク質,40Sリボソーム,RNA分解酵素や翻訳装置などが含まれる.RNA顆粒はRNA cargoの局在,安定性あるいは翻訳の調節を介して,遺伝子発現の転写後調節の鍵を握る構造であると認識されている.
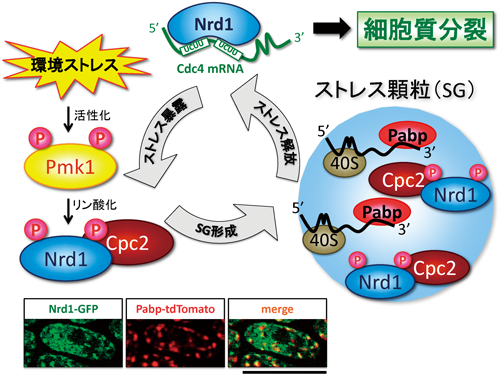
中でもSGはさまざまなストレス刺激(熱ショック,ヒ素,酸化ストレス,ウイルス感染,低酸素等)依存的に誘導され,mRNA,RNA結合タンパク質,40Sリボソームなどが構成要素として含まれる(図5).SGの形成はきわめてダイナミックかつ可逆的であり,ストレスに応答して刻々とその構成要素が変化をとげる.このようなSGが形成される意義は,ストレスに応答した翻訳制御やストレスに対する適応機構と考えられているが,その詳細な分子機構や構成因子,生理的役割のほとんどが謎である.
そこで我々は,Nrd1がTIA-1/TIAR同様にSGに局在する可能性について検証した.その結果,Nrd1の細胞内局在は熱ショックやヒ素などのストレス刺激に応答して細胞質内で凝集体を形成し,しかもその点状構造はSGのマーカーであるポリ(A)結合タンパク質(PABP)と共局在することが明らかになった30)(図5下の蛍光顕微鏡像).さらに,SGを誘導する各種ストレス刺激はPmk1 MAPKを活性化することから,Nrd1のSGへの移行とMAPKの活性化に機能的連関があるのではないか,との仮説を立てた.そして予想どおり,熱ショックやヒ素などのストレスは,Pmk1 MAPKの活性化と,それに引き続くNrd1のリン酸化とSGへの移行を引き起こすことがわかった30).MAPKによるNrd1のリン酸化レベルが高いほど,Nrd1のSG局在が顕著に誘導されることも明らかになった.
さらに興味深いことに,Nrd1を過剰発現すると,各種のストレス刺激が存在しない状況にも関わらずSGが形成され,しかもその効果は擬リン酸化型のNrd1DDでより顕著であった30).高等生物のTIA-1/TIARにおいても,過剰発現に伴い自らが凝集体を形成し,SGの核(seeds)としてSG形成を誘導,制御することが報告されている33).一方,Nrd1をノックアウトした細胞ではSGの形成が遅延し,一過性のストレスに感受性を示すことがわかった30).すなわち,Nrd1はMAPKによるリン酸化依存的にSGを誘導し,SGの形成と,ストレス応答機構において重要な役割を担うと考えられる.
それでは,一体どのようなメカニズムでNrd1はリン酸化依存的にSG形成を誘導するのであろうか? そこで,我々はNrd1の結合パートナーであるCpc2(高等生物のRACKホモログ)がNrd1のSG移行の鍵となるのではないか,と考えた.驚くべきことに,Nrd1はストレス依存的にCpc2と結合し,さらにCpc2ノックアウト細胞においてNrd1のヒ素刺激依存的かつ過剰発現依存的なSG移行/誘導が劇的に低下したことから,Nrd1とCpc2がストレス依存的に複合体を形成することによってSG形成のコアを形成するのではないか,という仮説を考えている30)(図5).
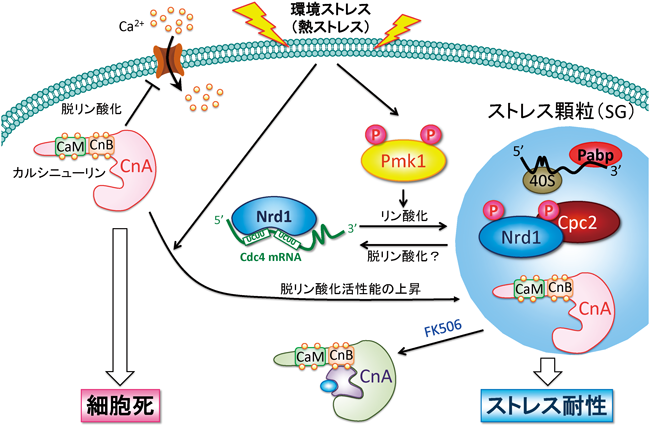
5. カルシニューリンは熱ショック依存的に活性化し,SGに移行する
それでは,SGを解離する仕組みは存在するのだろうか? また,MAPKによるリン酸化依存的なNrd1のSG制御機構に対して拮抗するような,Nrd1ホスファターゼは存在するのであろうか? そこで我々は,前述のカルシニューリンとPmk1 MAPKの拮抗的な機能的関係を説明しうる分子基盤の一つとして,カルシニューリンがNrd1の脱リン酸化酵素としてSG形成やストレス応答に関わる可能性を検証した.はじめに,熱刺激に応答したカルシニューリンの細胞内局在を解析した結果,カルシニューリンの局在は熱ストレスに応答して劇的に変化し,細胞質からドット状の構造体に移行した34).しかもこのカルシニューリン陽性な凝集体はPABPと共局在し,シクロヘキシミド処理により消失したことから,カルシニューリンはSGの構成因子であると考えられた34).そこで次に,熱ショックがカルシニューリンシグナルの活性化にどのような影響を与えるのかを調べる目的で,熱刺激に対しても安定したカルシニューリン活性測定を可能にするウミシイタケ(Renilla)ルシフェラーゼとカルシニューリン応答配列(CDRE)を融合したレポーター構築を確立した34).その結果,熱ショックを与えることにより,カルシニューリン活性は刺激時間依存的に上昇することが明らかになった.Ca2+以外にも高温ストレスという普遍的なストレス刺激に応答してカルシニューリンが活性化し,かつSGに移行することはきわめて興味深い.次に我々は,カルシニューリンシグナルの活性とSG移行という現象にどのような機能的関係があるのかを探る目的で,恒常的活性化型のCNΔC(カルシニューリンのC末端に存在するカルモジュリン結合部位を欠損させることにより,Ca2+シグナル非依存的な活性を獲得する8))を細胞内に発現させ,その局在を観察した.その結果,活性化型のCNΔCは野生型のカルシニューリンよりも顕著にSGに局在し,しかもそのSG局在はカルシニューリンの特異的阻害薬であるFK506処理により劇的に消失した34).これらの結果は,活性の高いカルシニューリンはSGに対する親和性が高く,かつカルシニューリンのSGへの移行にはホスファターゼ活性が必要であることを示唆している.
そこで,カルシニューリンがSG形成にどのような役割を果たしているのかを調べる目的で,カルシニューリンノックアウト(KO)細胞におけるSGの形成を解析した.そのため,前述のSGのマーカータンパク質であり,起点となるRNA結合タンパク質Nrd1を,内在性プロモーター下でGFP融合タンパク質として発現させ,正常細胞とカルシニューリンKO細胞における熱ショック後のNrd1のSGへの移行を解析した.すると,正常細胞においてはNrd1のSGへの移行がほとんど観察されない,きわめて短時間の熱刺激処理(42°C,5分)によって,カルシニューリンKO細胞ではNrd1がSGに移行することを確認した34).しかも,カルシニューリンKO細胞は,正常細胞よりも熱刺激に対して耐性を示すことが明らかとなった.この結果は,Nrd1 KO細胞が熱刺激に対して感受性を示し,かつSG形成が正常細胞に比べて遅延することと対照的である.これらの結果から,SGの形成は熱ショックを含めたストレスに生体が適応する上で重要な機構であること,さらにカルシニューリンはSG形成を負に制御する可能性が示唆された.
6. RNA顆粒の“シグナル伝達制御拠点”としての新たな役割
現在の仮説を図6に示す.通常の至的増殖環境ではカルシニューリンは細胞質あるいは細胞膜および中隔に局在し,Ca2+シグナルを速やかに細胞内に伝達する.すなわち,カルシニューリンは細胞質に局在する転写因子や細胞膜表面に局在するCa2+チャネルあるいは細胞質分裂のキーファクターなどの標的因子(基質)を迅速に脱リン酸化するのに適切な局在に配置されている.
一方,熱刺激によってカルシニューリンはSGへと移行し,しかもこの移行はカルシニューリンの酵素活性に依存する.この現象から,あたかもカルシニューリン活性がSG形成を誘導する(正に制御する)という解釈が成り立つ.ところが,予想に反して,カルシニューリンKO細胞ではNrd1などのSGのコアとなるタンパク質が,正常細胞よりも迅速にSGに移行することから,カルシニューリンはSG形成においてNrd1と拮抗するような負の調節を行っていることを示唆する.カルシニューリンKO細胞の示すストレス抵抗性という表現型もSGの負の制御因子としての役割にフィットする.
この一見相矛盾した現象の解釈として,カルシニューリンは酵素活性依存的にSGに“受動的に”移行し,隔離され,しかもSG形成を負に制御するのではないかと考えた.このように高い酵素活性を有するカルシニューリンがSG(あるいはその構成因子)に対して強い親和性を持つ生理的意義に関しては,SGが“シグナル伝達のハブ(制御拠点)”として機能し,ある閾値を超えて活性化したカルシニューリンを隔離することにより,過剰に活性化したCa2+/カルシニューリンシグナルの暴走を遮断し,細胞死などの望ましくない生体反応を回避するのではないか,と推測される(図6).実際にカルシニューリンを過剰発現すると,細胞増殖が顕著に阻害される.
このようなシグナル伝達制御拠点としてのSGの新たな概念と役割は,近年急速に注目を集めている.前述したように,RNA顆粒の主たる機能は主に「RNAの運命決定装置」として転写後の遺伝子発現調節の鍵を握ることである32).たとえばウイルスに感染したり異常タンパク質が蓄積したりすると,SGが形成され,非翻訳状態のmRNAを隔離することにより一時的にタンパク質合成が停止する.細胞がストレスから回復するとSGは消失し,翻訳が再開される.
一方,TakekawaらのグループよりSG形成とストレス応答MAPK経路制御の関わりが報告されて以来,翻訳制御,RNA代謝以外のSGの生理機能の解明に焦点が当てられた.Takekawaらは,ストレス応答MAP3KであるMTK1の結合分子としてRACK1を同定し,無刺激ではMTK1と細胞質で結合しているRACK1が,ヒ素などのストレス刺激に応答してMTK1と解離し,SGに移行することを発見した35).結果的に,活性化因子としてのRACK1を失ったストレス応答MAPK経路はその活性化が阻害されることとなる.このようなメカニズムは,SG形成によるストレス応答MAPK経路の活性化阻害とアポトーシス抑制機構として,きわめて興味深く,かつ種を超えて,TORやGSK3など重要なシグナル伝達分子がSGにおいて会合するという諸研究の端緒となった.
興味深いことに,SGは上記のようなシグナル伝達制御拠点としての役割に加えて,さまざまな疾患との関わりが浮かび上がりつつある.具体的には,がんやアルツハイマー病,筋委縮性側索硬化症(ALS)などの神経変性疾患との関わりが注目を集めている.たとえば上述のように,SGはストレスによるDNA損傷や異常タンパク質の蓄積を防ぎ,細胞損傷を回避するための防御機構と考えられてきた.つまり,細胞のエネルギーを緊急事態としての細胞損傷の修復や回復に必須な翻訳に専念させる仕組みと考えられる.したがって適切にSGを形成あるいは崩壊することが,翻訳の制御/抑制において重要であり,ひいてはウイルス感染や酸化ストレスに対する細胞の防御機構にも不可欠であると考えられている.実際に,高等生物においてもSGの形成不全がストレスに対する脆弱性を惹起したり,TIA-1ノックアウトマウスが重症の関節炎を発症したりするなど,SGはストレスや炎症に対して細胞レベルでも,生体レベルでも防御的な役割を担うことが明らかとなりつつある.
一方,ALSやアルツハイマー病などの神経変性疾患において,“病的な”SGが“持続的に”形成されていることが,これらの疾患の病態と密接に関わるという報告がある.特に,アルツハイマー病のモデルマウスおよびアルツハイマー病患者の脳においては,病態の進行に伴い,より顕著にSGが観察されることが報告されている.初期の段階では,TIA-1がリン酸化タウタンパク質と凝集体を形成しているが,さらに病状が進むと,リン酸化タウはRNA結合タンパク質TTP(tristetraprolin)と結合すると報告されている.さらに興味深いことに,TIA-1を過剰発現することにより,リン酸化タウの凝集体が形成され,互いに重なり合った局在を示す.これらの結果は,このように病的なSGが持続的に形成されることがリン酸化タウに随伴した病態と密接に関連していることを示唆している.
さらに一歩進んで,SG形成を神経変性疾患の治療標的とした研究もすでに開始されている.ハエや酵母などのモデル生物を用いて,ALSに伴う病的SGを改善するような遺伝子や化合物の探索が行われ36),GSK3β阻害剤(Glaxo)が同定されている.驚くべきことに,この阻害剤を投与されたハエは筋力を回復し,さらにラット由来の細胞においてもSG形成の阻害に効果的であった36).これらの結果は,SG形成機構が種を超えて保存されていることを示すのみならず,SGの機能異常を修復することが病態の改善に直結する例としてきわめて魅力的なものである.
冒頭にも述べたように,RNA顆粒形成の制御機構や生理機能はそのほとんどが謎に包まれている.今後さらに,RNA顆粒の構成因子や,その制御に関わるシグナル伝達機構の役割,さらにはRNA顆粒の“シグナルハブ”としての役割の解明が行われることにより,さまざまな疾患治療標的としてのRNA顆粒の重要性がいっそう注目を集めると考えられる.今回我々の発見したSGによるカルシニューリンの空間的制御機構が高等生物においても保存されているのであれば,アルツハイマー病や心疾患,ダウン症候群など,カルシニューリンシグナルの破綻に伴うさまざまな病態とRNA顆粒制御の関わりが明らかになることが期待される.RNA顆粒の制御機構あるいはその機能不全に伴う疾患の分子機構を明らかにする上で,酵母をはじめとしたモデル生物の貢献を願っている.
引用文献References
1) Stewart, A.-A., Ingebritsen, T.-S., Manalan, A., Klee, C.-B., & Cohen, P. (1982) FEBS Lett., 137, 80–84.
2) Liu, J., Farmer, J.-D. Jr., Lane, W.-S., Friedman, J., Weissman, I., & Schreiber, S.-L. (1991) Cell, 66, 807–815.
3) Clipstone, N.-A. & Crabtree, G.-R. (1992) Nature, 357, 695–697.
4) O’Keefe, S.-J., Tamura, J., Kincaid, R.-L., Tocci, M.-J., & O’Neill, E.-A. (1992) Nature, 357, 692–694.
5) Crabtree, G.-R. (2001) J. Biol. Chem., 276, 2313–2316.
6) McCaffrey, P.-G., Perrino, B.-A., Soderling, T.-R., & Rao, A. (1993) J. Biol. Chem., 268, 3747–3752.
7) Sugiura, R., Sio, S.-O., Shuntoh, H., & Kuno, T. (2002) Genes Cells, 7, 619–627.
8) Sugiura, R., Toda, T., Shuntoh, H., Yanagida, M., & Kuno, T. (1998) EMBO J., 17, 140–148.
9) Sugiura, R., Toda, T., Dhut, S., Shuntoh, H., & Kuno, T. (1999) Nature, 399, 479–483.
10) Sugiura, R., Kita, A., Shimizu, Y., Shuntoh, H., Sio, S.-O., & Kuno, T. (2003) Nature, 424, 961–965.
11) Ma, Y., Kuno, T., Kita, A., Asayama, Y., & Sugiura, R. (2006) Mol. Biol. Cell, 17, 5028–5037.
12) Doi, A., Kita, A., Kanda, Y., Uno, T., Asami, K., Satoh, R., Nakano, K., & Sugiura, R. (2015) Genes Cells, 20, 310–323.
13) Zhang, Y., Sugiura, R., Lu, Y., Asami, M., Maeda, T., Itoh, T., Takenawa, T., Shuntoh, H., & Kuno, T. (2000) J. Biol. Chem., 275, 35600–35606.
14) Yada, T., Sugiura, R., Kita, A., Itoh, Y., Lu, Y., Hong, Y., Kinoshita, T., Shuntoh, H., & Kuno, T. (2001) J. Biol. Chem., 276, 13579–13586.
15) Fujita, M., Sugiura, R., Lu, Y., Xu, L., Xia, Y., Shuntoh, H., & Kuno, T. (2002) Genetics, 161, 971–981.
16) Cheng, H., Sugiura, R., Wu, W., Fujita, M., Lu, Y., Sio, S.-O., Kawai, R., Takegawa, K., Shuntoh, H., & Kuno, T. (2002) Mol. Biol. Cell, 13, 2963–2676.
17) Lu, Y., Sugiura, R., Yada, T., Cheng, H., Sio, S.-O., Shuntoh, H., & Kuno, T. (2002) Genes Cells, 7, 1009–1019.
18) Kita, A., Sugiura, R., Shoji, H., He, Y., Deng, L., Lu, Y., Sio, S., Takegawa, K., Sakaue, M., Shuntoh, H., & Kuno, T. (2004) Mol. Biol. Cell, 15, 2920–2931.
19) He, Y., Sugiura, R., Ma, Y., Kita, A., Deng, L., Takegawa, K., Matsuoka, K., Shuntoh, H., & Kuno, T. (2006) Genes Cells, 11, 207–221.
20) Yu, Y., Kita, A., Udo, M., Katayama, Y., Shintani, M., Park, K., Hagihara, K., Umeda, N., & Sugiura, R. (2012) PLoS ONE, 7, e45324.
21) Kita, A., Higa, M., Doi, A., Satoh, R., & Sugiura, R. (2015) Biochem. Biophys. Res. Commun., 457, 273–279.
22) Higa, M., Kita, A., Hagihara, K., Kitai, Y., Doi, A., Nagasoko, R., Satoh, R., & Sugiura, R. (2015) Genes Cells, 20, 95–107.
23) Takahara, T. & Maeda, T. (2012) Mol. Cell, 47, 242–252.
24) Sio, S.-O., Suehiro, T., Sugiura, R., Takeuchi, M., Mukai, H., & Kuno, T. (2005) J. Biol. Chem., 280, 12231–12238.
25) 久能樹,杉浦麗子(2013)次世代がん戦略研究Updateがん基盤生物学—革新的シーズ育成に向けて—,pp.172–178, 南山堂.
26) Takada, H., Nishimura, M., Asayama, Y., Mannse, Y., Ishiwata, S., Kita, A., Doi, A., Nishida, A., Kai, N., Moriuchi, S., Tohda, H., Giga, H.-Y., Kuno, Y., & Sugiura, R. (2007) Mol. Biol. Cell, 18, 4794–4802.
27) Takada, H., Nishida, A., Domae, M., Kita, A., Yamano, Y., Uchida, A., Ishiwata, S., Fang, Y., Zhou, X., Masuko, T., Kinoshita, M., Kakehi, K., & Sugiura, R. (2010) Mol. Biol. Cell, 21, 674–685.
28) Ishiwata, S., Kuno, T., Takada, H., Koike, A., & Sugiura, R. (2008) Sourcebook of Models for Biomedical Research, pp.439–443, Humana Press Inc.
29) Satoh, R., Morita, T., Takada, H., Kita, A., Ishiwata, S., Doi, A., Hagihara, K., Taga, A., Matsumura, Y., Tohda, H., & Sugiura, R. (2009) Mol. Biol. Cell, 20, 2473–2485.
30) Satoh, R., Tanaka, A., Kita, A., Morita, T., Matsumura, Y., Umeda, N., Takada, M., Hayashi, S., Tani, T., Shinmyozu, K., & Sugiura, R. (2012) PLoS ONE, 7, e29683.
31) López de Silanes, I., Galbán, S., Martindale, J.-L., Yang, X., Mazan-Mamczarz, K., Indig, F.-E., Falco, G., Zhan, M., & Gorospe, M. (2005) Mol. Cell. Biol., 25, 9520–9531.
32) Anderson, P. & Kedersha, N. (2006) J. Cell Biol., 172, 803–808.
33) Kedersha, N.-L., Gupta, M., Li, W., Miller, I., & Anderson, P. (1999) J. Cell Biol., 147, 1431–1442.
34) Higa, M., Kita, A., Hagihara, K., Kitai, Y., Doi, A., Nagasoko, R., Satoh, R., & Sugiura, R. (2015) Genes Cells, 20, 95–107.
35) Arimoto, K., Fukuda, H., Imajoh, O.-S., Saito, H., & Takekawa, M. (2008) Nat. Cell Biol., 10, 1324–1332.
36) Kin, H.J., Raphael, A.R., Ladow, E.S., McGurk, L., Weber, R.A., Trojanowski, J.Q., Lee, V.M.-Y., Finkbeiner, S., Gitler, A.D., & Bonini, N.M. (2014) Nat. Genet., 46, 152–160.
著者紹介Author Profile
 佐藤 亮介(さとう りょうすけ)
佐藤 亮介(さとう りょうすけ)近畿大学薬学部創薬科学科助教.博士(薬学).
略歴1984年大阪府に生る.2007年近畿大学薬学部卒業.09年同大学院薬学研究科博士前期課程修了.12年同研究科博士後期課程修了.12年4月~14年3月微生物化学研究所(BIKAKEN)博士研究員,14年4月より現職.
研究テーマと抱負シグナル経路によって制御されるRNA結合タンパク質の機能解析を通して,RNA顆粒の未知なる生理機能を解明する.
ウェブサイトhttp://www.phar.kindai.ac.jp/genome
趣味フィッシング(詐欺ではない),サッカー・フットサル,ドライブ,ピアノ,ボウリングなど多趣味.
 杉浦 麗子(すぎうら れいこ)
杉浦 麗子(すぎうら れいこ)近畿大学薬学部創薬科学科教授,学科長.博士(医学).
略歴1992年神戸大学医学部卒業.精神科,内科の臨床研修を経て94年より神戸大学大学院医学系研究科入学.神戸大学医学部薬理学研究室助手,講師を経て2000年より同准教授.01年文部科学省在外研究員(英国王立癌研究所).04年より近畿大学薬学部教授.12年より現職.
研究テーマと抱負酵母モデル生物を用いたシグナル伝達とRNA制御機構の解明とゲノム創薬・疾患治療への応用.酵母遺伝学で得られた独創的な成果や命題を創薬やヒト疾患治療に応用できるか,という課題に挑戦中.〈そんな馬鹿げたアイデアは無理に決まっている〉と言われると燃えます.
ウェブサイトhttp://www.phar.kindai.ac.jp/genome
趣味読書,音楽鑑賞,酒(特に日本酒,ワイン)をたしなむこと,筋トレ.