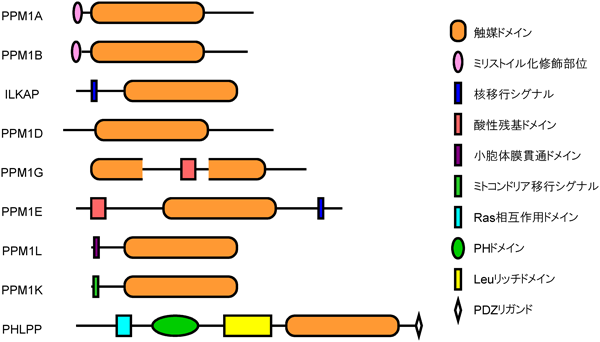
セリントレオニンホスファターゼは,PP1やPP2Aを代表とするPPP(phosphoprotein phosphatase)ファミリーと,それらとは異なる分子進化上の起源を持つPPM(metal-dependent protein phosphatase)ファミリーに大きく分類される[後者は以前,2C型プロテインホスファターゼ(PP2C)と呼ばれていたものであるが,新たな命名法によりPPMと呼称されるようになった].PPPファミリーのメンバーの大部分が,触媒サブユニットと調節サブユニットからなるオリゴマー酵素であるのに対し,PPMは基本的に単量体であり,構造学的には,触媒活性を担うホスファターゼドメインと,機能を規定するドメインが同一分子内に内蔵されているという特徴を持っている(図1).これまでに,哺乳動物細胞においては,17種類のPPM遺伝子産物の存在が報告されており,さまざまな細胞機能の制御に関与することが報告されている(表1).本稿では,本ファミリーの新たな機能とその制御メカニズムを最近の知見から紹介していきたい.なお,PPMファミリーのうち,PPM1Dの機能と制御に関しては,次稿に詳しいので本稿では取り上げない.
表1 哺乳動物のPPMファミリーメンバー| 名称 | 別名 | 関与する代表的な細胞機能 |
|---|
| PPM1A | PP2Cα | ストレス応答,TGFβシグナリング |
| PPM1B | PP2Cβ | ストレス応答,NF-κB経路 |
| PPM1D | Wip1 | 細胞周期のチェックポイント |
| PPM1E | CaMKP-N, POPX1 | CaMキナーゼ,細胞骨格 |
| PPM1F | CaMKP, POPX2 | CaMキナーゼ,細胞骨格 |
| PPM1G | PP2Cγ | RNAスプライシング,タンパク質の翻訳 |
| PPM1H | NERPP | 神経軸索の伸長,細胞周期 |
| PPM1J | PP2Cζ | ? |
| PPM1K | PP2Cκ | 分岐鎖アミノ酸の代謝 |
| PPM1L | PP2Cε | セラミド輸送,小胞体ストレス |
| PPM1M | PP2Cη | NF-κB経路 |
| ILKAP | PP2Cδ | RSK2経路 |
| PHLPP1 | SCOP | Akt, PKCおよびS6K |
| PHLPP2 | | Akt, PKCおよびS6K |
| PPTC7 | TA-PP2C | ? |
| PDP1 | | ピルビン酸脱水素酵素複合体 |
| PDP2 | | ピルビン酸脱水素酵素複合体 |
PPMという名称は,in vitroでの活性測定にMg2+やMn2+などの金属イオンの存在を必要とすることに由来している.最近,金属イオンに対する感受性の網羅的検索により,カドミウム(Cd2+)がPPMファミリーの強力な阻害物質(Ki=170~300 nM)として作用することが見いだされた1).PPMファミリーの活性中心は,βシートが並列に配置されたβサンドイッチ構造により形成され,2個の金属イオン(M1およびM2)がβストランドのC末端に位置する数個のアミノ酸残基に配位している.Cd2+はPPMの二つの金属結合部位のうち,M1と競合阻害することにより効果を示す.この発見は,Cd2+の汚染物質としてのさまざまな作用(発がん,免疫疾患,神経系の異常等)が,PPMの阻害により生じる可能性を示唆している.
1)がんの病態への関与
ⅰ)TGFβ経路の制御因子としてのPPM1Aとがん悪性化への関与
Smad2/3は,活性化されたtransforming growth factor β(TGFβ)受容体によりリン酸化を受けた後,co-SmadであるSmad4と複合体を形成して核移行し,さまざまな転写因子と相互作用することで遺伝子発現を制御する.その後,Smad2/3は核内で脱リン酸化され,RAN binding protein 3(RanBP3)が関与する機構により核外に移行される.PPM1AはTGFβ依存的に活性化されたSmad2/3を核内で脱リン酸化することにより,活性化型のSmad複合体の解体を促進し,Smad2/3の核外移行を促す2,3).Smad2/3の核外移行に関わるRanBP3はribosomal protein S6 kinase(RSK)またはAktによりリン酸化を受け,機能が抑制されているが,加えてPPM1Aはこのリン酸化部位をも脱リン酸化することにより,Smad2/3の核外移行を促進することも明らかとなった4).PPM1Aノックアウトマウスは,皮膚の創傷治癒の過程での上皮再形成に遅延が観察されたが,これは,創傷後のケラチノサイトの運動能が低下することに由来するものであり,Smad2/3のリン酸化の亢進と関連していた5).腫瘍形成においてTGFβシグナリング経路の活性化は,がん細胞の浸潤と転移を促進するとともに,上皮間葉転換(EMT)を亢進することによりがん悪性化の亢進に関わっていることが知られている.肝細胞がんの病理組織標本の観察から,肝細胞がんの進行につれて,PPM1Aの細胞内局在の核から細胞質への変化が観察され,それと逆相関するように,リン酸化型Smad2の核への蓄積が認められた6).一方,Gengらは膀胱がんの症例を検討し,PPM1Aの発現が低い症例では,筋層への高い浸潤がみられ,予後が悪いことを報告した.また,再発部位では原発巣に比較し,PPM1Aの発現量低下が顕著であり,PPM1Aの発現量とSmad2/3のリン酸化レベルに逆相関が認められた7).以上の臨床学的知見は,PPM1AがSmad2/3のリン酸化レベルの制御を介して腫瘍形成抑制因子として働くことを示している.
ⅱ)がん細胞の浸潤能におけるPPM1Fの役割
PAK(p21-activated kinase)はRhoファミリーGTPaseの下流で機能するシグナリング分子で,アクチン細胞骨格の構成,細胞運動性,アポトーシスの制御などに関わり,その脱抑制が乳がんや大腸がんをはじめとする多くのがんの病態と関連している.Kohらは,PAK結合性のグアニンヌクレオチド交換因子(PIX)と相互作用するタンパク質として,PPM1EおよびPPM1Fを同定した8).PPM1Fの発現は多くのがん細胞株で認められ,その発現レベルは浸潤能と相関関係を持っていた9).がん細胞株でのPPM1Fの過剰発現が浸潤能と運動性を高める一方で,PPM1Fの発現のノックダウンは運動性と浸潤能を抑制することが確認された.がん細胞株のPPM1Fのノックダウンは,β1インテグリンの発現レベルに影響を与え,その結果,ストレスファイバーと焦点接着(focal adhesion)の著しい減少を示した.以上の結果より,PPM1Fはβ1インテグリンの発現を介して細胞接着を制御し,浸潤能を亢進していることが示唆された.
ⅲ)PHLPP
PHLPPはPHLPP1およびPHLPP2の二つの遺伝子にコードされており,ホスファターゼドメインの他に,機能ドメインとしてRas相互作用ドメイン,pleckstrin homology(PH)ドメイン,leucine-rich repeatドメインおよびPDZドメイン結合モチーフを有する10).これまでに,Akt,PKC(protein kinase C)およびS6K(S6 kinase)の疎水性モチーフ内に位置する,活性化に必須なリン酸化部位を脱リン酸化し,キナーゼ活性の減弱(AktおよびS6K)やタンパク質の不安定化(PKC)を引き起こすことが報告されている11–13).
前立腺がんのゲノム解析の結果,PHLPP1およびPHLPP2遺伝子の欠失が,がん抑制遺伝子として著名なPTENの欠失頻度に匹敵するほどの高い頻度で起こっていることが明らかにされた14).PHLPP1およびPHLPP2の発現量低下は同様に大腸がんの臨床検体でも認められた15).初期段階の前立腺がんでは10%弱にPHLPP1遺伝子の欠失がみられたが,PTENとの共欠失はほとんど認められなかった.一方,転移相に至ったがん組織では,その約3割がPHLPP1欠損を有しており,ほとんどがPTEN,p53およびPHLPP2遺伝子も同時に欠失していた16).前立腺がん切除手術後の生存率も,腫瘍におけるPHLPP/PTENの発現レベルと有意な相関関係を示した.一方,PHLPP1遺伝子が単独で欠損したマウスは,腫瘍を形成しなかったが,PTENのヘテロ欠損を持ち,さらにp53遺伝子に変異が生じると高頻度に前立腺に腫瘍を形成した16).以上のヒトおよびマウスでの解析結果から,PHLPP1およびPTENの欠失により惹起されたAktの活性化が,いったんは細胞のセネッセンス(細胞老化)を誘導しがん化への移行を抑制するが,変異導入によりp53が活性を失うと,セネッセンスが解除され腫瘍形成に移行するというモデルが提唱された.また,PHLPP1の欠損は,一時的にPHLPP2の発現を増加させPHLPP1の欠損を補償するというメカニズムが備わっていることも明らかになった.
2)細胞周期
トラスツズマブ(trastuzumab)はヒトがん遺伝子HER2(c-erbB-2)の遺伝子産物であるHER2タンパク質に特異的に結合することで抗腫瘍効果を発揮する抗がん剤であるが,治療の過程で薬剤抵抗性を生じることが問題となっている.この薬剤抵抗性を招く原因として,PTENの欠損やPI3キナーゼの活性型突然変異によるPI3キナーゼ/Akt経路の活性化が知られている.Lee-HoeflichらはsiRNAライブラリーを用いて新規のトラスツズマブ抵抗性遺伝子の検索を行い,PPM1Hを同定した17).PPM1Hは,脳神経系において高い発現がみられ,中枢神経系における神経再生能に関わる分子として同定されていたが,本研究によりCDK(cyclin-dependent kinase)インヒビターであるp27のThr187を脱リン酸化し安定化することが明らかになった.PPM1Hの発現が低下しているがん細胞では,p27のリン酸化レベルの上昇とタンパクの不安定化が観察された.また,乳がん患者においてPPM1Hの発現レベルの低下と,予後の間に相関関係があることが観察された.以上の結果より,PPM1Hが細胞周期の制御を介してがん抑制遺伝子として機能していることが明らかとなった.
3)小胞体における機能
PPM1Lは,ストレス応答シグナル伝達経路の制御因子として我々が初めて同定し,当初プロテインホスファターゼ2Cε(PP2Cε)と命名したものである18,19).その後,N末端に膜貫通ドメインを持ち,細胞質側に触媒ドメインを配した形で小胞体(endoplasmic reticulum: ER)膜に局在するという,セリントレオニンホスファターゼとしては,大変ユニークな存在様式を持っていることを明らかにした20).形質膜上の主要なリン脂質であるスフィンゴミエリン(sphingomyelin: SM)は,ER膜上でセラミドとして合成された後,ゴルジ体に輸送されSMに変換されるが,このERからゴルジへの輸送を担うのが脂質輸送体CERT(ceramide transfer protein)である21).CERTは基底状態では,SRモチーフと呼ばれる領域が多重リン酸化されることにより不活性な状態で細胞質にとどめられているが,セラミド輸送が必要になる局面でER-ゴルジ体間にリクルートされ,そこで脱リン酸化を受け活性化される22).我々は,PPM1LがER膜上の足場タンパク質であるVAPAと会合することを見いだし,FFATモチーフを介してリクルートされたCERTを,脱リン酸化し活性化することを明らかにした20).
一方,Luらは,PPM1LがERストレスセンサーの一つであるIRE1の制御に関わることを報告した23).ERストレスのシグナルは,IRE1,PERKおよびATF6の3種類のセンサーで感知された後,細胞質・核へと伝達されていく.そのうち,IRE1の活性化には,ERストレスに応答したセンサーの二量体化とそれに伴うSer724を含む複数の残基の分子間自己リン酸化が必要である.Luらは,PPM1Lが他のERストレス応答経路に影響を与えることなく,IRE1のSer724のみを特異的に脱リン酸化することを明らかにした.
以上のように,PPM1LがERの機能の重要な制御因子であることが明らかにされる一方,2種類の系統マウスを用いて行った量的形質座位(QTL)解析の結果,PPM1Lが肥満形成に関する遺伝子として同定された24).ERの機能の制御が,肥満形成にどのように関わっているのかについては,今後の興味深い研究課題といえる.
4)AMPK
AMP活性化プロテインキナーゼ(AMP-activated protein kinase: AMPK)は酵母からヒトまで高度に保存されているセリントレオニンキナーゼで,細胞内のエネルギー状態のセンサーとしてエネルギー恒常性の維持において重要な役割を担っている.活性化には触媒サブユニット(AMPKα)のThr172のリン酸化が必要であるが,これまで,このリン酸化を担うプロテインキナーゼについてはLKB1とCaMKKβ(calcium-calmodulin-dependent protein kinase kinase β)が主要な分子であることが明らかにされていた.しかし,脱リン酸化を担うホスファターゼについては,PPMタイプのホスファターゼが機能するという報告があるのみで詳細は不明であった.Steimbergらは,TNFα(tumor necrosis factor α)がインスリン抵抗性を誘導するメカニズムの解析の過程で,TNFα依存性に転写活性化されたPPM1Aが,AMPKを脱リン酸化し不活性化することを報告した25).一方,Vossらは,2型糖尿病治療薬であるビグアナイドへの感受性という観点から,AMPKの脱リン酸化を担うホスファターゼを検索した.細胞をビグアナイドの一つであるフェンホルミンで処理すると,AMPKのリン酸化レベルおよびキナーゼ活性が上昇することが明らかにされていたが,彼らは,その際,細胞内のPPM型ホスファターゼの酵素活性が低下することを見いだした.個別のアイソフォームへの影響を調べた結果,フェンホルミン処理によるPPM活性の減弱は,PPM1EとPPM1Fが阻害を受けた結果であることが判明した.さらに,siRNAを用いた発現のノックダウン実験およびAMPKαのアイソフォームとの会合実験より,PPM1EがAMPKαの二つのアイソフォームのうちAMPKα2を特異的に脱リン酸化することを明らかにした26).また,我々は,HeLa細胞内におけるLKB1依存性のAMPK活性化への個々のPPMファミリーメンバーの共発現の影響を検討することにより,PPM1BもAMPKαの脱リン酸化を担うことを見いだした27).PPM1Bは,PPM1Aとアミノ酸配列レベルで75%の相同性を有する類縁のアイソフォームではあるが,細胞内局在が異なっている(PPM1A:核局在,PPM1B:細胞質).このことは,PPM1AとPPM1BがAMPK複合体を細胞内の異なった場所で脱リン酸化することを示唆している.また,PPM1EがAMPKα2を特異的に脱リン酸化するのに対し,PPM1AとPPM1AはAMPKα1とAMPKα2を同等に脱リン酸化する27).以上の結果は,それぞれのアイソフォームが細胞内の異なった部位で,異なったAMPK複合体に機能することを示しており,エネルギー代謝制御における役割の特異性を示唆している.
PPPファミリーに属するPP1やPP2Aは,触媒サブユニットと調節サブユニットの組み合わせによりダイナミックな制御を受けていることがよく知られている.一方,PPMファミリーは基本的に単量体として機能することもあって,その調節メカニズムの実体に関しては不明な点が多く残されている.本節では,PPMファミリーの制御メカニズムについて,現在までに報告された例を紹介したい.
1)リン酸化
ⅰ)JNKによるPPM1Jの機能制御
ストレス応答性プロテインキナーゼ(stress activated protein kinase: SAPK)経路は,プロテインキナーゼであるMKKK,MKKおよびSAPK(JNKおよびp38)による三段階のリン酸化反応からなる細胞内情報伝達系で,環境ストレスや炎症性サイトカインなどの細胞外刺激に応答して活性化され,細胞の生存や炎症反応などさまざまな細胞機能を制御している.これまで,PPMファミリーの複数のメンバーがSAPK経路の制御因子として機能することが明らかにされてきた28).我々は,PPMファミリーのメンバーが,フィードバックメカニズムによりSAPKで制御される可能性を考え,各メンバーのJNKおよびp38によるin vitroでのリン酸化を試みた.その結果,PPM1JがJNKにより特異的にリン酸化を受けることが明らかとなった.PPM1Jは,精巣に特異的に発現を示す分子として同定されたもので,SUMO化修飾との関連が指摘されている29).リン酸化部位はSer92とThr205の2か所で,いずれの部位も細胞内でストレス依存性にリン酸化レベルが増強し,特に,Ser92のリン酸化により,PPM1Jのホスファターゼ活性が抑制された30).興味深いことに,これらのリン酸化部位はPPM1MとPPM1Hでも保存されており,これらのアイソフォームがSAPK以外のプロテインキナーゼにより制御される可能性を示唆した.
ⅱ)PHLPP
PHLPP1は半減期4時間の比較的不安定なタンパク質である.Liらは,この不安定性にSCF E3リガーゼの基質結合サブユニットであるβ-TrCPを介したユビキチン化およびそれに続くプロテオソーム依存性のタンパク質分解が関与することを明らかにした31).一般に,β-TrCPを介した分解は,ターゲットとなるタンパク質のリン酸化が引き金となって開始されるが,PHLPP1においては,ホスファターゼドメイン内の4か所のセリンまたはトレオニン残基がCK1(casein kinase 1)およびGSK3β(glycogen synthase kinase β)によりリン酸化され,β-TrCPによる認識部位(phospho-degron)を形成していた.細胞の非刺激時には,活性化型(脱リン酸化型)のGSK3βがPHLPP1をリン酸化しβ-TrCPとの結合を促進することで,PHLPP1を不安定化しているが,増殖刺激によるAktの活性化に伴いGSK3βがリン酸化され非活性化型に転換されるとPHLPP1が脱リン酸化型になり,プロテオソーム依存性分解から逃れることで安定化する.興味深いことに,直腸がんの臨床検体においてβ-TrCPの発現亢進が認められ,これが直腸がんにおけるPHLPP1発現量低下の原因である可能性が指摘された.
ⅲ)PPM1E
PPM1Eは,培養細胞を用いた研究成果より,核局在のホスファターゼと考えられていた.ところが,ラット脳のPPM1Eの大部分が細胞質に存在しており,細胞質局在型のPPM1Eは核移行シグナルを含むC末端領域を欠いていることが報告された32).阻害剤を用いた研究により,このC末端領域の切断は,ユビキチン/プロテオソーム系により起こることが明らかとなった33).C末端領域の限定分解は,ホスファターゼ活性を上昇させるとともに,細胞内局在を核から細胞質に変化させ,それに伴って,核に存在するCaMキナーゼ(CaMK)IV(CaMKIV)から細胞質に存在するCaMKIを脱リン酸化するようになった.一方,プロセッシングを受け細胞質に局在するようになったPPM1Eは,CaMKIによりSer550がリン酸化され,触媒活性が亢進する34).PPM1EはCaMKIだけでなく,CaMKIIも基質とすることから,PPM1Eの活性化を介したCaMKIによるCaMKIIの制御機構の存在が示唆された.
2)酸化還元状態による活性の調節
Babaらは,PPM1Eのシステイン残基が分子内ジスルフィド結合形成により架橋されており,この架橋が活性を抑制することを見いだした35).この架橋は還元剤により解消され,それに伴って酵素活性も上昇した.チロシンホスファターゼにおいては,活性酸素種が触媒活性の制御において,重要な役割を担うことが明らかにされていたが,セリントレオニンホスファターゼにおいても,同様なメカニズムが存在することが示唆された.
3)脂質修飾
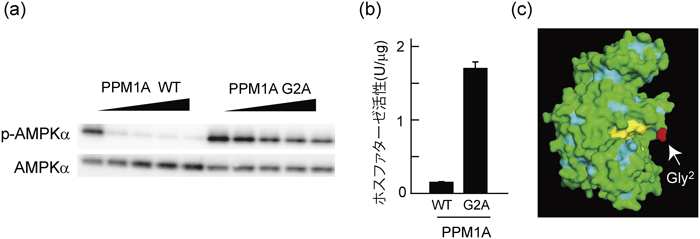
タンパク質のN-ミリストイル化はN末端のグリシンにミリスチン酸が付加する脂質修飾であり,タンパク質–脂質相互作用やタンパク質–タンパク質相互作用において重要な役割を担っている.今回我々は,PPM1AとPPM1BがN-ミリストイル化されており,この修飾が基質認識に重要な役割を担うことを明らかにした27).PPM1AとPPM1BのG2A変異体は,細胞内で生理的基質であるAMPKやSAPKを脱リン酸化できなかった.リン酸化AMPKを基質としたin vitroの反応系で,これらのG2A変異体は,野生型と比較して触媒活性が減弱していた.興味深いことに,G2A変異体のホスファターゼ活性は,人工的な化合物であるp-ニトロフェニルリン酸を基質とした場合は逆に亢進していた.立体構造上,N末端のグリシンが活性中心の近傍に位置することを勘案すると,N末端に結合したミリストイル基は活性中心の構造を安定化し,正確な基質特異性を獲得するのに貢献していると考えられた(図2).
4)相互作用分子による機能調節
我々は,ゴルジ体に局在するacyl-CoA-binding domain-containing protein 3(ACBD3)がPPM1LにGoldドメインを介して会合し,ゴルジ体膜近傍にPPM1Lをリクルートすることを見いだした36).前述のようにPPM1LはER膜とゴルジ体膜が近接する部位でVAPA依存性にCERTを脱リン酸化し,リン脂質の輸機能を亢進するが,ACBD3は,PPM1Lをゴルジ体との近接部にリクルートすることにより,CERTを効率よく脱リン酸化させると考えられる.
過去のセリントレオニンホスファターゼの研究の歴史を振り返ってみると,有効な阻害剤の存在(たとえばオカダ酸やサイクロスポリンAなど)がin vivoでの機能解明に重要な役割を演じてきた.その点で,特異的な阻害剤が利用できなかったことが,PPMファミリーの機能的研究の進展が遅れた一つの理由となっていた.しかし,siRNAをはじめとする分子細胞生物学的な手法の進歩により,PPMファミリーのさまざまな特異的な機能が次々と明らかとなってきた.特に,最近ではがんをはじめとする種々の疾患との関連が明らかにされつつあり,創薬の対象として,新たな治療薬の開発に結びつくものと期待される.一方,PPMファミリーの制御機構については,いまだ不明な点が多く,今後の研究の進展がのぞまれる.
引用文献References
1) Pan, C., Liu, H.D., Gong, Z., Yu, X., Hou, X.B., Xie, D.D., Zhu, X.B., Li, H.W., Tang, J.Y., Xu, Y.F., Yu, J.Q., Zhang, L.Y., Fang, H., Xiao, K.H., Chen, Y.G., Wang, J.Y., Pang, Q., Chen, W., & Sun, J.P. (2013) Sci. Rep., 3, 2333.
2) Lin, X., Duan, X., Liang, Y.Y., Su, Y., Wrighton, K.H., Long, J., Hu, M., Davis, C.M., Wang, J., Brunicardi, F.C., Shi, Y., Chen, Y.G., Meng, A., & Feng, X.H. (2006) Cell, 125, 915–928.
3) Bourgeois, B., Gilquin, B., Tellier-Lebegue, C., Ostlund, C., Wu, W., Perez, J., El Hage, P., Lallemand, F., Worman, H.J., & Zinn-Justin, S. (2013) Sci. Signal., 6, ra49.
4) Dai, F., Shen, T., Li, Z., Lin, X., & Feng, X.H. (2011) EMBO Rep., 12, 1175–1181.
5) Yang, X., Teng, Y., Hou, N., Fan, X., Cheng, X., Li, J., Wang, L., Wang, Y., Wu, X., & Yang, X. (2011) J. Biol. Chem., 286, 42267–42273.
6) Wu, S.K., Wang, B.J., Yang, Y., Feng, X.H., Zhao, X.P., & Yang, D.L. (2007) World J. Gastroenterol., 13, 4554–4559.
7) Geng, J., Fan, J., Ouyang, Q., Zhang, X., Yu, J., Xu, Z., Li, Q., Yao, X., Liu, X., & Zheng, J. (2014) Oncotarget, 5, 5700–5711.
8) Koh, C.G., Tan, E.J., Manser, E., & Lim, L. (2002) Curr. Biol., 12, 317–321.
9) Susila, A., Chan, H., Loh, A.X., Phang, H.Q., Wong, E.T., Tergaonkar, V., & Koh, C.G. (2010) Cell Cycle, 9, 179–187.
10) Newton, A.C. & Trotman, L.C. (2014) Annu. Rev. Pharmacol. Toxicol., 54, 537–558.
11) Gao, T., Furnari, F., & Newton, A.C. (2005) Mol. Cell, 18, 13–24.
12) Gao, T., Brognard, J., & Newton, A.C. (2008) J. Biol. Chem., 283, 6300–6311.
13) Liu, J., Stevens, P.D., Li, X., Schmidt, M.D., & Gao, T. (2011) Mol. Cell. Biol., 31, 4917–4927.
14) Taylor, B.S., Schultz, N., Hieronymus, H., Gopalan, A., Xiao, Y., Carver, B.S., Arora, V.K., Kaushik, P., Cerami, E., Reva, B., Antipin, Y., Mitsiades, N., Landers, T., Dolgalev, I., Major, J.E., Wilson, M., Socci, N.D., Lash, A.E., Heguy, A., Eastham, J.A., Scher, H.I., Reuter, V.E., Scardino, P.T., Sander, C., Sawyers, C.L., & Gerald, W.L. (2010) Cancer Cell, 18, 11–22.
15) Liu, J., Weiss, H.L., Rychahou, P., Jackson, L.N., Evers, B.M., & Gao, T. (2009) Oncogene, 28, 994–1004.
16) Chen, M., Pratt, C.P., Zeeman, M.E., Schultz, N., Taylor, B.S., O’Neill, A., Castillo-Martin, M., Nowak, D.G., Naguib, A., Grace, D.M., Murn, J., Navin, N., Atwal, G.S., Sander, C., Gerald, W.L., Cordon-Cardo, C., Newton, A.C., Carver, B.S., & Trotman, L.C. (2011) Cancer Cell, 20, 173–186.
17) Lee-Hoeflich, S.T., Pham, T.Q., Dowbenko, D., Munroe, X., Lee, J., Li, L., Zhou, W., Haverty, P.M., Pujara, K., Stinson, J., Chan, S.M., Eastham-Anderson, J., Pandita, A., Seshagiri, S., Hoeflich, K.P., Turashvili, G., Gelmon, K.A., Aparicio, S.A., Davis, D.P., Sliwkowski, M.X., & Stern, H.M. (2011) Cancer Discov., 1, 326–337.
18) Li, M.G., Katsura, K., Nomiyama, H., Komaki, K., Ninomiya-Tsuji, J., Matsumoto, K., Kobayashi, T., & Tamura, S. (2003) J. Biol. Chem., 278, 12013–12021.
19) Saito, J., Toriumi, S., Awano, K., Ichijo, H., Sasaki, K., Kobayashi, T., & Tamura, S. (2007) Biochem. J., 405, 591–596.
20) Saito, S., Matsui, H., Kawano, M., Kumagai, K., Tomishige, N., Hanada, K., Echigo, S., Tamura, S., & Kobayashi, T. (2008) J. Biol. Chem., 283, 6584–6593.
21) Hanada, K., Kumagai, K., Yasuda, S., Miura, Y., Kawano, M., Fukasawa, M., & Nishijima, M. (2003) Nature, 426, 803–809.
22) Kumagai, K., Kawano, M., Shinkai-Ouchi, F., Nishijima, M., & Hanada, K. (2007) J. Biol. Chem., 282, 17758–17766.
23) Lu, G., Ota, A., Ren, S., Franklin, S., Rau, C.D., Ping, P., Lane, T.F., Zhou, Z.H., Reue, K., Lusis, A.J., Vondriska, T., & Wang, Y. (2013) Mol. Metab., 2, 405–416.
24) Chen, Y., Zhu, J., Lum, P.Y., Yang, X., Pinto, S., MacNeil, D.J., Zhang, C., Lamb, J., Edwards, S., Sieberts, S.K., Leonardson, A., Castellini, L.W., Wang, S., Champy, M.F., Zhang, B., Emilsson, V., Doss, S., Ghazalpour, A., Horvath, S., Drake, T.A., Lusis, A.J., & Schadt, E.E. (2008) Nature, 452, 429–435.
25) Steinberg, G.R., Michell, B.J., van Denderen, B.J., Watt, M.J., Carey, A.L., Fam, B.C., Andrikopoulos, S., Proietto, J., Gorgun, C.Z., Carling, D., Hotamisligil, G.S., Febbraio, M.A., Kay, T.W., & Kemp, B.E. (2006) Cell Metab., 4, 465–474.
26) Voss, M., Paterson, J., Kelsall, I.R., Martin-Granados, C., Hastie, C.J., Peggie, M.W., & Cohen, P.T. (2011) Cell. Signal., 23, 114–124.
27) Chida, T., Ando, M., Matsuki, T., Masu, Y., Nagaura, Y., Takano-Yamamoto, T., Tamura, S., & Kobayashi, T. (2013) Biochem. J., 449, 741–749.
28) Tamura, S., Toriumi, S., Saito, J., Awano, K., Kudo, T.A., & Kobayashi, T. (2006) Cancer Sci., 97, 563–567.
29) Kashiwaba, M., Katsura, K., Ohnishi, M., Sasaki, M., Tanaka, H., Nishimune, Y., Kobayashi, T., & Tamura, S. (2003) FEBS Lett., 538, 197–202.
30) Awano, K., Amano, K., Nagaura, Y., Kanno, S., Echigo, S., Tamura, S., & Kobayashi, T. (2008) Biochemistry (Mosc), 47, 7248–7255.
31) Li, X., Liu, J., & Gao, T. (2009) Mol. Cell. Biol., 29, 6192–6205.
32) Kitani, T., Okuno, S., Nakamura, Y., Tokuno, H., Takeuchi, M., & Fujisawa, H. (2006) J. Neurochem., 96, 374–384.
33) Sueyoshi, N., Nimura, T., Onouchi, T., Baba, H., Takenaka, S., Ishida, A., & Kameshita, I. (2012) Arch. Biochem. Biophys., 517, 43–52.
34) Onouchi, T., Sueyoshi, N., Ishida, A., & Kameshita, I. (2012) Biochem. Biophys. Res. Commun., 422, 703–709.
35) Baba, H., Sueyoshi, N., Shigeri, Y., Ishida, A., & Kameshita, I. (2012) Arch. Biochem. Biophys., 526, 9–15.
36) Shinoda, Y., Fujita, K., Saito, S., Matsui, H., Kanto, Y., Nagaura, Y., Fukunaga, K., Tamura, S., & Kobayashi, T. (2012) FEBS Lett., 586, 3024–3029.
著者紹介Author Profile
 小林 孝安(こばやし たかやす)
小林 孝安(こばやし たかやす)東北大学加齢医学研究所准教授.医学博士.
略歴1985年東北大学農学部卒業.91年同大学院医学研究科博士課程修了.同年東北大学抗酸菌病研究所助手.2007年東北大学加齢医学研究所助教.09年より現職.
研究テーマと抱負PPMファミリーの機能解析.特に,PPMファミリーのメンバーの機能がいかに制御されているかに興味を持って研究を行っています.
趣味歩くこと.