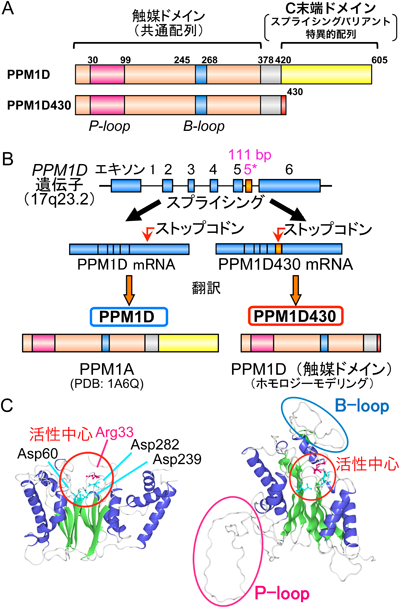
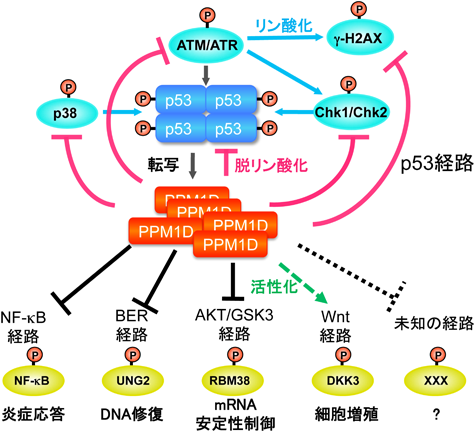
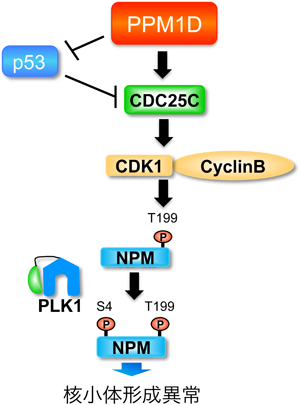
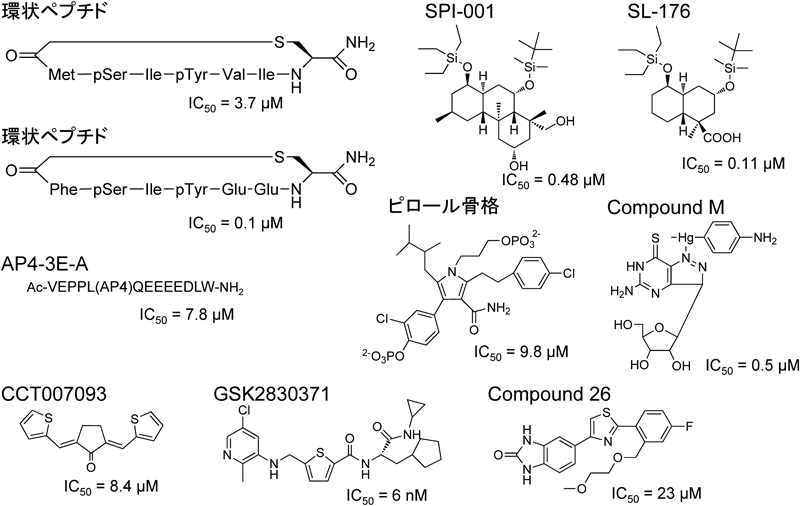
3) Das, A.K., Helps, N.R., Cohen, P.T., & Barford, D. (1996) EMBO J., 15, 6798–6809.
6) Cerami, E., Gao, J., Dogrusoz, U., Gross, B.E., Sumer, S.O., Aksoy, B.A., Jacobsen, A., Byrne, C.J., Heuer, M.L., Larsson, E., Antipin, Y., Reva, B., Goldberg, A.P., Sander, C., & Schultz, N. (2012) Cancer Discov., 2, 401–404.
7) Gao, J., Aksoy, B.A., Dogrusoz, U., Dresdner, G., Gross, B., Sumer, S.O., Sun, Y., Jacobsen, A., Sinha, R., Larsson, E., Cerami, E., Sander, C., & Schultz, N. (2013) Sci. Signal., 6, pl1.
11) Rayter, S., Elliott, R., Travers, J., Rowlands, M.G., Richardson, T.B., Boxall, K., Jones, K., Linardopoulos, S., Workman, P., Aherne, W., Lord, C.J., & Ashworth, A. (2008) Oncogene, 27, 1036–1044.
12) Kleiblova, P., Shaltiel, I.A., Benada, J., Sevcik, J., Pechackova, S., Pohlreich, P., Voest, E.E., Dundr, P., Bartek, J., Kleibl, Z., Medema, R.H., & Macurek, L. (2013) J. Cell Biol., 201, 511–521.
13) Zhang, L., Chen, L.H., Wan, H., Yang, R., Wang, Z., Feng, J., Yang, S., Jones, S., Wang, S., Zhou, W., Zhu, H., Killela, P.J., Zhang, J., Wu, Z., Li, G., Hao, S., Wang, Y., Webb, J.B., Friedman, H.S., Friedman, A.H., McLendon, R.E., He, Y., Reitman, Z.J., Bigner, D.D., & Yan, H. (2014) Nat. Genet., 46, 726–730.
14) Ruark, E., Snape, K., Humburg, P., Loveday, C., Bajrami, I., Brough, R., Rodrigues, D.N., Renwick, A., Seal, S., Ramsay, E., Duarte Sdel, V., Rivas, M.A., Warren-Perry, M., Zachariou, A., Campion-Flora, A., Hanks, S., Murray, A., Ansari Pour, N., Douglas, J., Gregory, L., Rimmer, A., Walker, N.M., Yang, T.P., Adlard, J.W., Barwell, J., Berg, J., Brady, A.F., Brewer, C., Brice, G., Chapman, C., Cook, J., Davidson, R., Donaldson, A., Douglas, F., Eccles, D., Evans, D.G., Greenhalgh, L., Henderson, A., Izatt, L., Kumar, A., Lalloo, F., Miedzybrodzka, Z., Morrison, P.J., Paterson, J., Porteous, M., Rogers, M.T., Shanley, S., Walker, L., Gore, M., Houlston, R., Brown, M.A., Caufield, M.J., Deloukas, P., McCarthy, M.I., Todd, J.A., Breast, Ovarian Cancer Susceptibility, C., Wellcome Trust Case Control, C., Turnbull, C., Reis-Filho, J.S., Ashworth, A., Antoniou, A.C., Lord, C.J., Donnelly, P., & Rahman, N. (2013) Nature, 493, 406–410.
20) Natrajan, R., Lambros, M.B., Rodriguez-Pinilla, S.M., Moreno-Bueno, G., Tan, D.S., Marchio, C., Vatcheva, R., Rayter, S., Mahler-Araujo, B., Fulford, L.G., Hungermann, D., Mackay, A., Grigoriadis, A., Fenwick, K., Tamber, N., Hardisson, D., Tutt, A., Palacios, J., Lord, C.J., Buerger, H., Ashworth, A., & Reis-Filho, J.S. (2009) Clin. Cancer Res., 15, 2711–2722.
34) Gilmartin, A.G., Faitg, T.H., Richter, M., Groy, A., Seefeld, M.A., Darcy, M.G., Peng, X., Federowicz, K., Yang, J., Zhang, S.Y., Minthorn, E., Jaworski, J.P., Schaber, M., Martens, S., McNulty, D.E., Sinnamon, R.H., Zhang, H., Kirkpatrick, R.B., Nevins, N., Cui, G., Pietrak, B., Diaz, E., Jones, A., Brandt, M., Schwartz, B., Heerding, D.A., & Kumar, R. (2014) Nat. Chem. Biol., 10, 181–187.
35) Cheeseman, M.D., Faisal, A., Rayter, S., Barbeau, O.R., Kalusa, A., Westlake, M., Burke, R., Swan, M., van Montfort, R., Linardopoulos, S., & Jones, K. (2014) Bioorg. Med. Chem. Lett., 24, 3469–3474.
41) Zhang, L., Liu, L., He, Z., Li, G., Liu, J., Song, Z., Jin, H., Rudolph, K.L., Yang, H., Mao, Y., Zhang, L., Zhang, H., Xiao, Z., & Ju, Z. (2015) Hepatology, 61, 2030–2041.