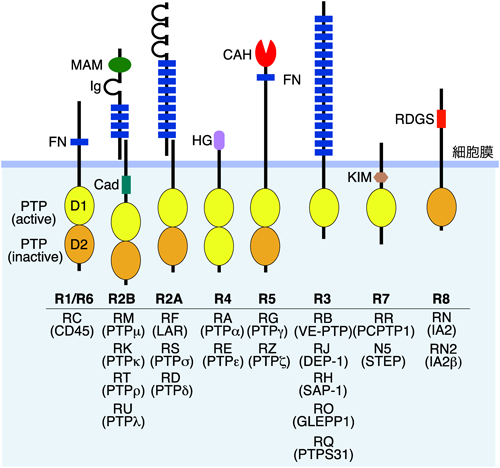
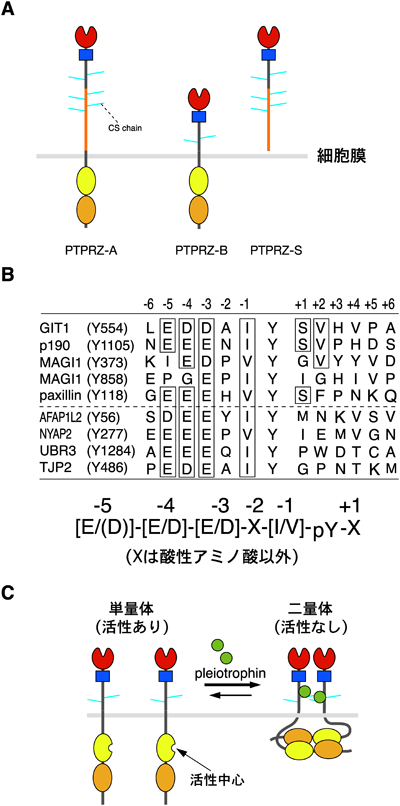
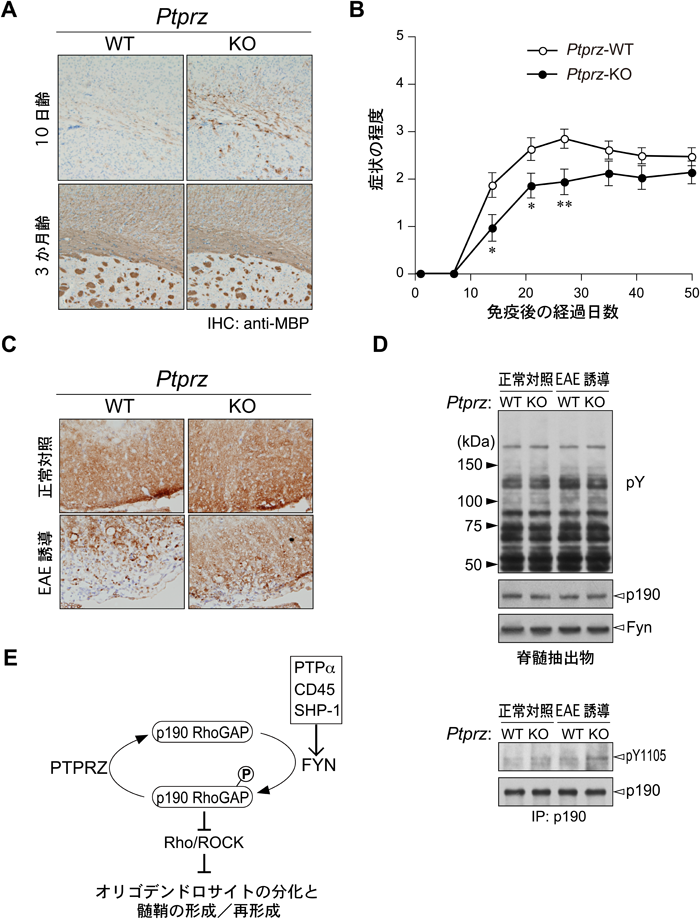
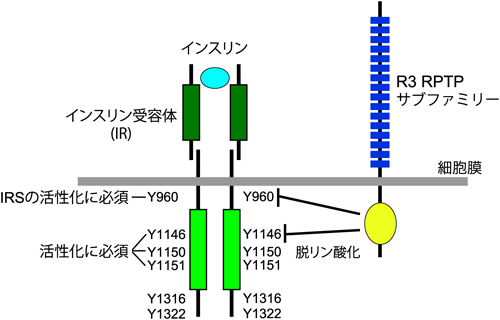
2) Kawachi, H., Tamura, H., Watakabe, I., Shintani, T., Maeda, N., & Noda, M. (1999) Brain Res. Mol. Brain Res., 72, 47–54.
4) Chow, J.P., Fujikawa, A., Shimizu, H., Suzuki, R., & Noda, M. (2008) J. Biol. Chem., 283, 30879–30889.
5) Chow, J.P., Fujikawa, A., Shimizu, H., & Noda, M. (2008) Neurosci. Lett., 442, 208–212.
6) Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J.W., & Maffei, L. (2002) Science, 298, 1248–1251.
7) Gogolla, N., Caroni, P., Lüthi, A., & Herry, C. (2009) Science, 325, 1258–1261.
8) Bradbury, E.J., Moon, L.D., Popat, R.J., King, V.R., Bennett, G.S., Patel, P.N., Fawcett, J.W., & McMahon, S.B. (2002) Nature, 416, 636–640.
9) Ishikawa, Y., Imagama, S., Ohgomori, T., Ishiguro, N., & Kadomatsu, K. (2015) Neurosci. Lett., 593, 13–18.
10) Shintani, T., Watanabe, E., Maeda, N., & Noda, M. (1998) Neurosci. Lett., 247, 135–138.
11) Fujikawa, A., Shirasaka, D., Yamamoto, S., Ota, H., Yahiro, K., Fukada, M., Shintani, T., Wada, A., Aoyama, N., Hirayama, T., Fukamachi, H., & Noda, M. (2003) Nat. Genet., 33, 375–381.
12) Müller, S., Kunkel, P., Lamszus, K., Ulbricht, U., Lorente, G.A., Nelson, A.M., von Schack, D., Chin, D.J., Lohr, S.C., Westphal, M., & Melcher, T. (2003) Oncogene, 22, 6661–6668.
13) Ulbricht, U., Eckerich, C., Fillbrandt, R., Westphal, M., & Lamszus, K. (2006) J. Neurochem., 98, 1497–1506.
14) Harroch, S., Palmeri, M., Rosenbluth, J., Custer, A., Okigaki, M., Shrager, P., Blum, M., Buxbaum, J.D., & Schlessinger, J. (2000) Mol. Cell. Biol., 20, 7706–7715.
15) Lafont, D., Adage, T., Gréco, B., & Zaratin, P. (2009) Behav. Brain Res., 201, 29–40.
16) Niisato, K., Fujikawa, A., Komai, S., Shintani, T., Watanabe, E., Sakaguchi, G., Katsuura, G., Manabe, T., & Noda, M. (2005) J. Neurosci., 25, 1081–1088.
17) Tamura, H., Fukada, M., Fujikawa, A., & Noda, M. (2006) Neurosci. Lett., 399, 33–38.
18) Fujikawa, A., Matsumoto, M., Kuboyama, K., Suzuki, R., & Noda, M. (2015) PLoS ONE, 10, e0119361.
19) Kawachi, H., Fujikawa, A., Maeda, N., & Noda, M. (2001) Proc. Natl. Acad. Sci. USA, 98, 6593–6598.
20) Fukada, M., Kawachi, H., Fujikawa, A., & Noda, M. (2005) Methods, 35, 54–63.
21) Fujikawa, A., Fukada, M., Makioka, Y., Suzuki, R., Chow, J.P., Matsumoto, M., & Noda, M. (2011) J. Biol. Chem., 286, 37137–37146.
22) Fujikawa, A., Chow, J.P., Shimizu, H., Fukada, M., Suzuki, R., & Noda, M. (2007) J. Biochem., 142, 343–350.
24) Meng, K., Rodriguez-Pena, A., Dimitrov, T., Chen, W., Yamin, M., Noda, M., & Deuel, T.F. (2000) Proc. Natl. Acad. Sci. USA, 97, 2603–2608.
25) Ratcliffe, C.F., Qu, Y., McCormick, K.A., Tibbs, V.C., Dixon, J.E., Scheuer, T., & Catterall, W.A. (2000) Nat. Neurosci., 3, 437–444.
27) Desai, D.M., Sap, J., Schlessinger, J., & Weiss, A. (1993) Cell, 73, 541–554.
28) Jiang, G., den Hertog, J., Su, J., Noel, J., Sap, J., & Hunter, T. (1999) Nature, 401, 606–610.
29) Nam, H.J., Poy, F., Krueger, N.X., Saito, H., & Frederick, C.A. (1999) Cell, 97, 449–457.
30) Nam, H.J., Poy, F., Saito, H., & Frederick, C.A. (2005) J. Exp. Med., 201, 441–452.
31) Maeda, N., Nishiwaki, T., Shintani, T., Hamanaka, H., & Noda, M. (1996) J. Biol. Chem., 271, 21446–21452.
33) Maeda, N., Ichihara-Tanaka, K., Kimura, T., Kadomatsu, K., Muramatsu, T., & Noda, M. (1999) J. Biol. Chem., 274, 12474–12479.
34) Fukada, M., Fujikawa, A., Chow, J.P.H., Ikematsu, S., Sakuma, S., & Noda, M. (2006) FEBS Lett., 580, 4051–4056.
35) Barr, A.J., Ugochukwu, E., Lee, W.H., King, O.N., Filippakopoulos, P., Alfano, I., Savitsky, P., Burgess-Brown, N.A., Müller, S., & Knapp, S. (2009) Cell, 136, 352–363.
36) Nandi, S., Cioce, M., Yeung, Y.G., Nieves, E., Tesfa, L., Lin, H., Hsu, A.W., Halenbeck, R., Cheng, H.Y., Gokhan, S., Mehler, M.F., & Stanley, E.R. (2003) J. Biol. Chem., 288, 21972–21986.
37) Najm, F.J., Madhavan, M., Zaremba, A., Shick, E., Karl, R.T., Factor, D.C., Miller, T.E., Nevin, Z.S., Kantor, C., Sargent, A., Quick, K.L., Schlatzer, D.M., Tang, H., Papoian, R., Brimacombe, K.R., Shen, M., Boxer, M.B., Jadhav, A., Robinson, A.P., Podojil, J.R., Miller, S.D., Miller, R.H., & Tesar, P.J. (2015) Nature, 522, 216–220.
38) Kuboyama, K., Fujikawa, A., Masumura, M., Suzuki, R., Matsumoto, M., & Noda, M. (2012) PLoS ONE, 7, e48797.
39) Kuboyama, K., Fujikawa, A., Suzuki, R., & Noda, M. (2015) J. Neurosci., 35, 12162–12171.
40) Wolf, R.M., Wilkes, J.J., Chao, M.V., & Resh, M.D. (2001) J. Neurobiol., 49, 62–78.
41) Oganesian, A., Poot, M., Daum, G., Coats, S.A., Wright, M.B., Seifert, R.A., & Bowen-Pope, D.F. (2003) Proc. Natl. Acad. Sci. USA, 100, 7563–7568.
42) Shintani, T., Ihara, M., Sakuta, H., Takahashi, H., Watakabe, I., & Noda, M. (2006) Nat. Neurosci., 9, 761–769.
43) Sakuraba, J., Shintani, T., Tani, S., & Noda, M. (2013) J. Biol. Chem., 32, 23421–23431.
44) Shintani, T., Higashi, S., Takeuchi, Y., Gaudio, E., Trapasso, F., Fusco, A., & Noda, M. (2015) J. Biochem., in press.
46) Krishnan, N., Koveal, D., Miller, D.H., Xue, B., Akshinthala, S.D., Kragelj, J., Jensen, M.R., Gauss, C.M., Page, R., Blackledge, M., Muthuswamy, S.K., Peti, W., & Tonks, N.K. (2014) Nat. Chem. Biol., 10, 558–566.