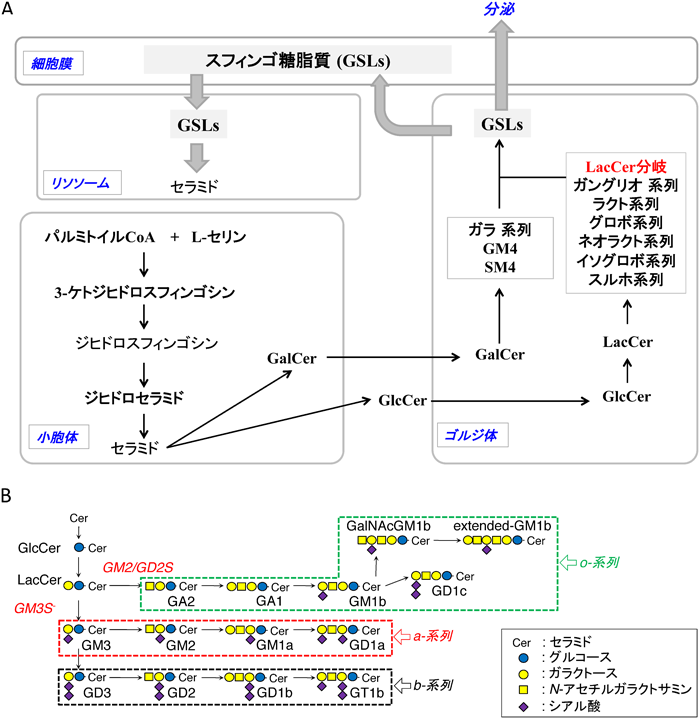
ドイツ生まれの神経化学者であるJ. L. W. Thudichum(1829~1901)は,ヒトの脳の両親媒性の不思議な脳物質に対して,ギリシャ神話のスフィンクスにちなんで謎の脂質,すなわち,「スフィンゴシン」と名づけた1).長鎖アミノアルコールであるスフィンゴシン(スフィンゴイド塩基)に脂肪酸が結合し,セラミドにリン酸基を有するものをスフィンゴリン脂質と呼び,スフィンゴミエリンは動物細胞膜のスフィンゴ脂質の主要成分である.一方,糖が結合したものをスフィンゴ糖脂質と呼ぶ.スフィンゴ糖脂質は細胞膜脂質二重層の外側に発現し,セラミドにグルコース,ガラクトース,N-アセチルガラクトサミン,N-アセチルグルコサミン,シアル酸などの糖や硫酸が段階的に酵素付加することによって,実に多様な分子が生合成される(図1).スフィンゴ糖脂質の構造多様性は,ラクトシルセラミド(LacCer)に作用する六つの酵素の発現が律速段階である(図1A).本小論の中心であるガングリオシドはシアル酸を含むスフィンゴ糖脂質である.ガングリオシドファミリー生合成の最初の分子は,ラクトシルセラミドにシアル酸が転移して生成するGM3である.そもそもGM3は,糖脂質研究の草分けである山川民夫博士がウマ赤血球からヘマトシドとして同定されたものである1).私にガングリオシド研究のきっかけを与えてくれたのは,グルコシルセラミド合成酵素の阻害剤(D-PDMP)を開発したこと2),そしてこのGM3合成酵素(GM3S, ST3GAL5)の遺伝子クローニングに参画したことである3).図1Bに示すように,GM3からは多様なガングリオシドが生合成されるが,これらは,細胞や組織において特徴的な発現特異性を示す.現在までに,GM3Sをはじめとして,種々の合成酵素のノックアウトマウス(KOマウス)の解析や,ガングリオシド合成酵素および分解酵素欠損症の研究から,細胞種特異的に発現するガングリオシド分子種の機能が見いだされつつある4,5).筆者は,このように多様な構造のガングリオシドを細胞特異的に発現する生命システムを必然的に獲得したのかという問いに対する答えを求め続けている5).本稿では,筆者の30年にわたる研究の軌跡を顧みて,ガングリオシドの今後を探ってみたい.
2. グルコシルセラミド合成酵素阻害剤D-threo-PDMP(D-PDMP)の開発
筆者のスフィンゴ糖脂質の研究は,1985年,ジョンズ・ホプキンス大学の岸本安夫教授の紹介で,ミシガン大学のノーマン・レーディン教授の研究室(Neuroscience laboratory building, Mental Health Research Institute)への留学に始まる.与えられたテーマは,グルコシルセラミド合成酵素の阻害物質の合成である.レーディン教授は,「セラミドの類似体である1-phenyl-2-decanoylamino-3-morpholino-1-propanol(PDMP)という化合物はグルコシルセラミド合成酵素を阻害する可能性があるが,PDMPの四つの光学異性体のどれに阻害活性があるか確かめてくれ」と光学分割の参考書を渡された.当時は光学分割用担体の開発が始まったばかりで,現在のように光学分割カラムで分取できるわけではない.まず,PDMPを数回のバッチで約30グラム合成し,クロロホルム/エーテルで再結晶したところ,比較的簡単にジアステレオマー(DL-erythroとDL-threo体)が分離できた.両者のグルコシルセラミド合成酵素阻害活性を調べたところ,DL-threo体のみに活性があった.天然型セラミドはD-erythro(2S, 3R)であることから,この結果は,私もレーディン教授も予想外であり,信州大医学部の武富教授の研究室から私の来る数年前にレーディン研に留学されていた原先生にNMRでの確認をお願いし,ようやくその立体構造を確信することができた.次は,いよいよさまざまなカウンターアシッドを用いてのエナンチオマーの分離である.くる日も来る日も得られた結晶を(1R)-(−)-カンファン酸クロリドで誘導化し,薄層クロマトグラフィーで光学異性体の分離を確認すること約半年が過ぎようとしていた.ミシガンの厳しい冬の土曜日の朝,DL-threo体と当モルのジベンゾイル-D-酒石酸をアセトン溶媒中,スターラーでゆっくり撹拌していたところ,フラスコの中でキラキラと冬の日光を反射している結晶を見たときのことは今でもはっきり覚えている.グルコシルセラミド合成酵素阻害物質D-threo-PDMPの誕生の瞬間である2).ミシガン大学から帰国した私はその後,生化学工業の山本龍人氏と水谷當稱社長にお会いしたことがきっかけで,東京研究所探索研究部門において,スフィンゴ糖脂質生合成制御物質についての研究を展開した.D-threo-PDMPは広く糖脂質研究者の実験に用いられ6–9),現在米国では,D-threo-PDMPの経口投与可能な類縁体をゴーシェ病の治療薬として用いた臨床治験が進んでいる10,11).さらに,糖尿病12),多発性嚢胞腎(PKD)13),脂肪肝14)などのモデル動物で有効性が見いだされている.表1にグルコシルセラミド合成酵素阻害剤としてD-threo-PDMPおよびPDMP類縁体およびデオキシノジリマイシン誘導体の構造についてまとめた.詳細については総説を参照されたい15).
表1 グルコシルセラミド合成酵素阻害剤 | 構造式 | IC50[µM] | 培養細胞での使用濃度[µM] |
|---|
| PDMP類縁体 | D-threo-PDMP | 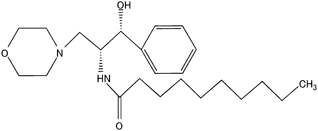 | 5 | 5~20 |
| D-threo-PPPP (P4)63) | 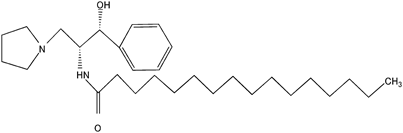 | 0.5 | 5~20 |
| Ethylenedioxy-P464) | 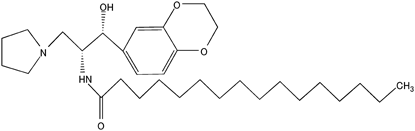 | 0.1 | 0.01~0.1 |
| D-threo-PBPP65) | 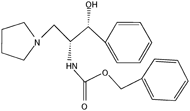 | 0.3 | 5~160 |
| デオキシノジリマイシン(dNM)誘導体 | N-butyl-dNM (Migulstat)15) | 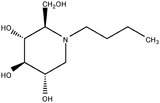 | 20 | 5~500 |
| Adamantan-pentyl-dNM15) | 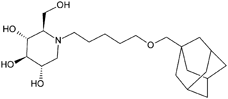 | 0.025 | 0.01~0.1 |
一方,D-threo-PDMPのエナンチオマーであるL-threo-PDMPには,ガングリオシド生合成促進効果が認められ16,17),神経突起進展活性18),シナプス機能および長期増強促進作用19),脳虚血モデルラットにおける再灌流障害抑制作用20)など,神経機能賦活効果を見いだした21).L-threo-PDMPのガングリオシド生合成促進効果は,酵素源として細胞破砕液を使用した場合には認められず,細胞レベルでのみ活性を示す.そこで,L-threo-PDMPの結合タンパク質を探索したが,ガングリオシド生合成促進効果のメカニズムは不明のままである.
生化学工業東京研究所時代(1991~1998年)には,永井克孝先生がグループディレクターとして理化学研究所国際フロンティア研究システム糖鎖機能研究を展開されていた.永井先生や東京都臨床医学総合研究所の鈴木明身先生には,示唆に富むさまざまなアドバイスを頂戴し,日本の糖脂質研究の伝統をご教示いただいた.永井先生は昨年ご逝去されたが,先生の哲学とお言葉を昨日のことのように思い出している.また,シアトルのワシントン大学の箱守仙一郎先生や五十嵐靖之先生(現北海道大学)との共同研究を始めさせていただいたのもこの時期である.箱守先生には,本学分子生体膜研究所顧問としてご指導いただいている.
1998年,箱守仙一郎教授の研究室から北海道大学薬学部に赴任された五十嵐靖之教授の研究室の助教授となった私は,五十嵐研究室の立ち上げとともに,ここまでの研究から生命科学者としてのオリジナリティーを築く自負をこめて未来への可能性を模索していた.当時,ヒト全ゲノム解読がなされ,スフィンゴ糖脂質の生合成酵素群の遺伝子クローニング競争の真っただ中であった.そのようなときに,当時自治医科大学におられた斎藤雅樹教授が行っていた数百種類の多様なスフィンゴ糖脂質の中のシアル酸を含むガングリオ系ガングリオシドファミリー生合成の律速酵素であるGM3合成酵素(GM3S)の遺伝子クローニングプロジェクトに参画することができ,幸運にも世界の先陣を切ることができた.このプロジェクトに参画できたのは,福岡大学薬学部助手時代(永松淳雄教授)に私が3LLルイス肺がん由来細胞株より,GM3を発現していないクローンであるJ5株についての研究をしていたことからである22).このGM3欠損J5株にヒト脳cDNAライブラリーを導入し,世界初のGM3S発現クローニングに成功した3).
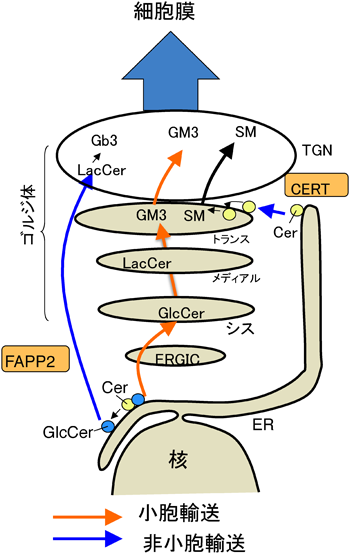
この仕事がきっかけで,GM3Sの分子生物学を開始することになった.細胞内におけるスフィンゴ脂質の生合成制御機構について,現時点での作業仮説も含めて図2に示した.国立感染症研究所の花田賢太郎博士は,ceramide traffickingにちなんでCERTと呼ばれる小胞体からゴルジ体へ特異的にセラミドを運ぶ分子を発見した23).CERTに結合したセラミドは,CERTのPHドメインとゴルジ膜局所に濃縮されたホスファチジルイノシトールと小胞体・ゴルジ体オルガネラ部位を形成し,主としてスフィンゴミエリン(SM)の生合成に利用される.また,小胞体内腔で合成されたセラミドの一部は,小胞体膜およびシスゴルジ膜の細胞質側グルコシルセラミド(GlcCer)になる.GlcCerは,Golgi-associated four-phosphate adaptor proteinのFAPP2と結合し,トランスゴルジに輸送され,ゴルジ体膜の内側にフリップした後,ラクトシルセラミド(LacCer)に生合成されることが示された24,25).おそらく,LacCer合成酵素はゴルジ体で局在が異なる2種類が存在し,FAPP2と結合したGlcCerはLacCerのグロボ系などの中性糖脂質(Gb3など)の生合成に利用されるのであろう.一方,LacCer合成酵素はGM3合成酵素(GM3S)と複合体を形成してガングリオシドファミリーの合成を制御している可能性が報告されている26).したがって,細胞特異的なスフィンゴ糖脂質分子種の発現機構の包括的な理解には種々の生合成酵素の細胞内局在および動態を徹底的に解明していくことが不可欠である.
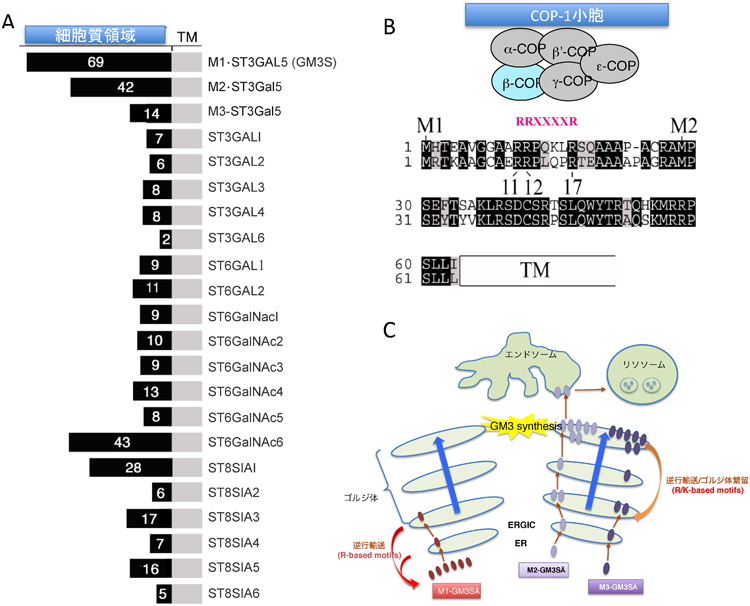
それでは,GM3以降の複合ガングリオシドの生合成はどのように制御されているのであろうか? 当研究室の博士研究員であった上村聡志君は,いくつかのガングリオシド生合成酵素で細胞質領域のN末端の長さが異なるアイソフォームを見いだし,その意義について検討していた.彼は,GM3SにはN末端側の細胞質領域の長さが異なる3種類のアイソフォーム(M1-, M2-, M3-GM3S)が存在し,それぞれの細胞内動態が大きく異なることを明らかにした27).注目されるのは,M1-GM3Sは細胞外領域が68アミノ酸あり,シアル酸転移酵素ファミリーの中でも最も長いことである(図3A).GM3Sの三つのアイソフォームは,二つのmRNAバリアント(mSAT-IaとmSAT-Ib)からleaky scanningにより産生される27).leaky scanningとは,最初のメチオニン(AUG)が開始コドンとしてリボソームに認識されず,さらに下流のメチオニンが開始コドンとして認識されて,翻訳が開始することである.開始コドンの認識には,アデニンから3塩基上流の塩基がアデニン,もしくはグアニンから4塩基下流の塩基がグアニンであることが重要であると考えられている.そのため,leaky scanningはこの開始コドン認識配列が十分でないときに起こり,GM3Sの場合も開始コドンの前をリボソームによる認識に最適だとされるGCC ACC(コザック配列)にした場合,このleaky scanningが大幅に抑制される.これらアイソフォーム間の性質の違いで,最も興味深いことは,細胞質領域の最も長いM1-GM3SのみがN末端側に逆行輸送シグナルR-basedモチーフを持ち,小胞体に局在することである(図3B).M1-GM3Sの逆行輸送は不完全であり,一部のM1-GM3Sは逆行輸送を逃れ,ゴルジ体まで運ばれた後,GM3を合成する.つまり,M1-GM3Sは安定して小胞体に局在しながら,ゆっくりとゴルジ体へGM3Sを供給していることが示唆される.また,M2-GM3SとM3-GM3Sはどちらもゴルジ体に局在しているが,M2-GM3Sは速やかにリソソームへ運ばれて分解されるのに対し,M3-GM3Sは安定してゴルジ体に繋留する.3種類のアイソフォームの量的バランスと局在のバランスがGM3合成の制御に重要であると推測している(図3C)28).GM3SはⅡ型の膜タンパク質であり複数のアスパラギン結合型糖鎖が付加される.我々は,これらの糖鎖が酵素活性に必須であり,立体構造の一部分を担っていることを示唆した29).さらに,M2-GM3SやM3-GM3Sには,現段階では,明確なシグナル配列が見つかっていないことから,未知のメカニズムでゴルジ体に繋留,もしくはリソソームへの輸送が行われていると考えられる.このように,3種類のアイソフォームが存在する生理的意義について,不明な点は残されているが,少なくとも3種類のアイソフォームが異なるメカニズムでゴルジ体に輸送・繋留していることは明らかである27,28).今後,ラクトシルセラミドに作用する6種の酵素(図1A)の細胞内動態が解明され,スフィンゴ糖脂質生合成制御機構の全貌が解明されることを期待している.
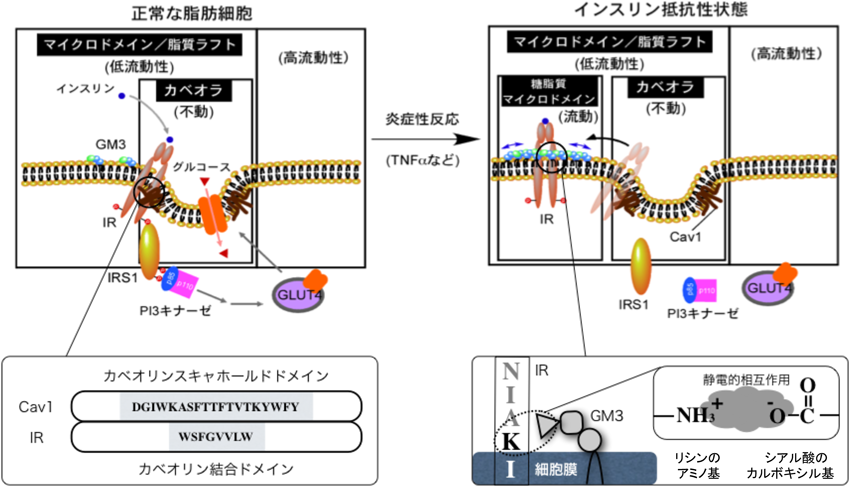
4. マイクロドメイン病としてのインスリン抵抗性の提唱
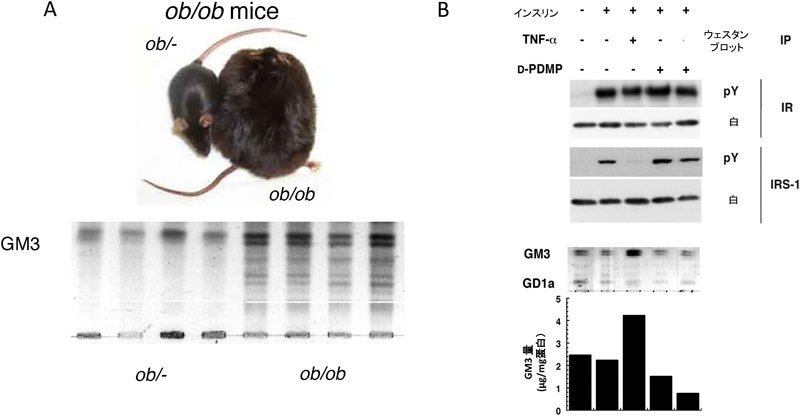
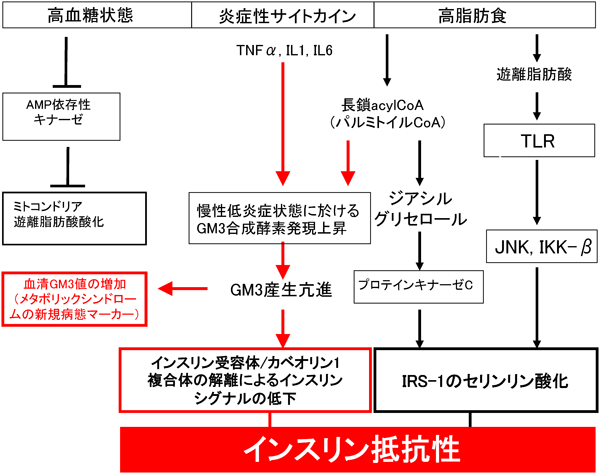
肥満糖尿病モデルであるdb/db,ob/obマウスやZucker fa/faラットなどの肥満モデル動物の脂肪組織では,それらのコントロール動物(lean)と比較すると腫瘍壊死因子α(TNFα)の著しい産生上昇が認められる30).これらの副睾丸脂肪組織のGM3合成酵素遺伝子発現を解析したところ,GM3S mRNAおよびGM3レベルの顕著な上昇が認められ,脂肪細胞のインスリン抵抗性にGM3の発現上昇が関与している可能性が示された(図4A)31).GM3の増加は,高脂肪食を負荷し高血糖を誘導したC57BL6マウスの内臓脂肪組織においても確認された32).脂肪細胞では,スフィンゴ糖脂質,スフィンゴミエリン,コレステロールなどのラフト脂質が集積して形成される細胞膜カベオラマイクロドメインにインスリン受容体(IR)が存在していることから,細胞膜におけるインスリンシグナル伝達においてもこれらの脂質環境場が影響を与えている可能性がある.TNFα刺激によりインスリン抵抗性を獲得した3T3-L1脂肪細胞のラフト脂質組成を調べると,GM3が特異的に2~3倍増加していた(図4B)31).そこで,グルコシルセラミド合成酵素の特異的阻害剤D-PDMP2)を用いて,TNFα刺激で増加したGM3の発現を抑制すると,IRS-1(insulin receptor substrate-1)のチロシンリン酸化の抑制がほぼ解除されたことから,GM3の異常な発現上昇がインスリン抵抗性を惹起していることが強く示唆された(図4B)31).さらに,樺山一哉博士(現大阪大学准教授)は蛍光標識IRおよびカベオリン-1の生細胞イメージング(FRAP法),免疫沈降法,およびミラノ大学Sandro Sonnino教授が派遣してくれたNicoletta Loberto博士と行った光感受性GM3を用いた架橋実験などにより,GM3の発現上昇によってIR βサブユニットの細胞膜直上のリシン残基とGM3のシアル酸残基の静電的相互作用が増大した結果,IR/カベオリン-1複合体が解離する分子機構を証明した(図5)33).これらの研究から「2型糖尿病などの生活習慣病は,スフィンゴ糖脂質の発現異常によってマイクロドメインの構成・構造および機能が変化し,シグナル伝達が異常になったマイクロドメイン病である」という病態概念を提唱している5,34–37).生活習慣病研究分野でのスフィンゴ脂質/糖脂質研究は急速に進みつつあり,創薬や新たな診断法の開発において今後大きく発展することが期待される.図6に我々が提唱する新たなインスリン抵抗性発症機序を示す.
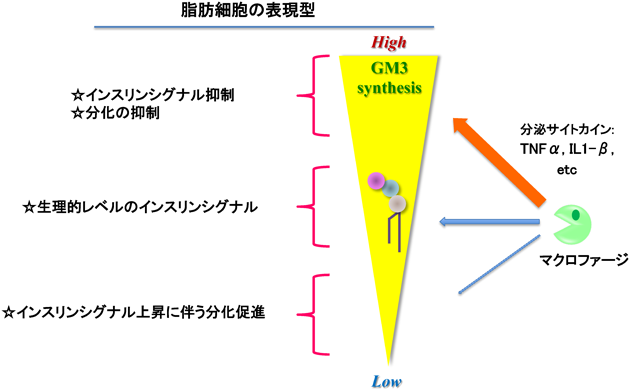
腸間膜脂肪組織より得られたstromal vascular(間質血管細胞群)画分に存在する前駆脂肪細胞の成熟脂肪細胞への分化は,常在性マクロファージによって制御されていることが知られている38).常在性マクロファージを除去すると前駆脂肪細胞の分化速度が促進されるとともに,インスリンシグナルも亢進していた32).驚くべきことに,この常在性マクロファージを除去した前駆脂肪細胞および分化誘導した脂肪細胞ではGM3を含むスフィンゴ糖脂質の発現が遺伝子レベルから抑制されており,定常状態においても脂肪細胞のスフィンゴ糖脂質の発現は,常在性マクロファージから分泌されている液性因子により制御されていることを明らかにした32).さらに,GM3S KOマウス胎仔から得られた線維芽細胞(MEF)は脂肪細胞への分化速度が対照のMEFに比較して速く,インスリンシグナルも亢進していた32).これらの結果より,GM3およびGM3から生合成されるガングリオシド分子種は,生体の恒常性維持およびメタボリックシンドロームにおける慢性炎症状態において,インスリンシグナルを制御する生理活性脂質としての働きが示された(図7)39).
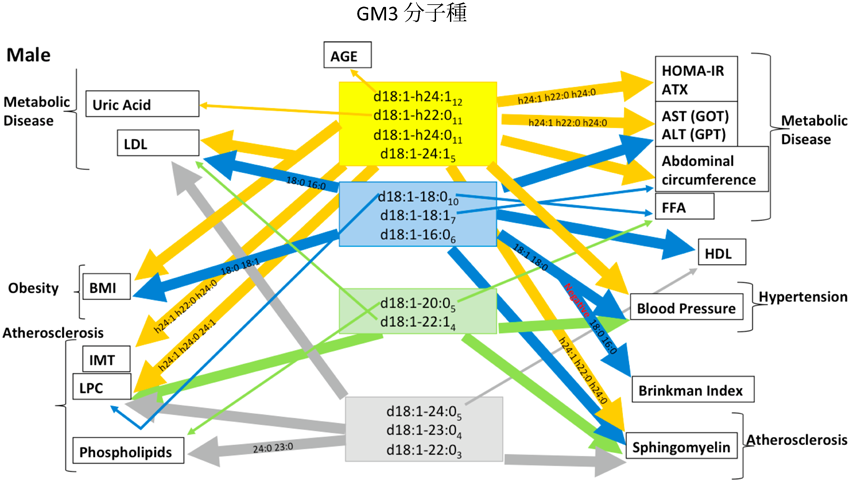
メタボリックシンドローム(内臓脂肪症候群)は内臓脂肪の蓄積を基盤として,ひとりに複数の危険因子が集中し,動脈硬化性疾患(心筋梗塞や脳梗塞など)の危険性を高める複合型リスク症候群状態である40).現在のところ,メタボリックシンドロームの主要な危険因子であるインスリン抵抗性を簡便に診断する指標がない.血液中の血球成分を除いた血漿または血清中には,GM3を主成分として,GD3,GD1a,GM2およびGT1bなどが存在していることが知られている41).また,血漿または血清中のガングリオシド量は,自己免疫疾患や胃がんで増加傾向が認められることが報告されているが,肥満およびインスリン抵抗性状態における脂肪細胞または脂肪組織のGM3発現の増加が血液試料において検知しうるか否かはまったく不明であった.ヒト血清中の主要なガングリオシドもGM3であり,健常人では約5 µg/mLの濃度で存在している41).我々は糖尿病などのメタボリックシンドローム患者血清のGM3値が上昇すること42),さらに,GM3の分子種を東海大学糖鎖科学研究所の鈴木明身教授の協力を得てLC-MS/MSで調べたところ,GM3のセラミド部分のアシル鎖2位の水酸基付加体(d18:1,OH24:0やd18:1,OH24:1など)が特異的に増加していることを見いだした(図8)43).この水酸化されたGM3分子種は2型糖尿病をはじめとする複雑なメタボリックシンドロームの慢性低炎症病態を新たな角度から検出することができる新規な病態マーカーである可能性があり,病態との詳細な相関関係を現在検証中である.
5. ガングリオシド分子種選択的なT細胞サブセットの機能制御
T細胞の抗原認識機構およびT細胞受容体(TCR)シグナル伝達機構は非常によく研究されてきた分野である.近年,顕微鏡による生細胞の可視化技術の向上もあって,T細胞と抗原提示細胞APCがその接触部分に形成する“免疫シナプス”の動的変化も明らかにされてきた44).これまでにT細胞活性化におけるガングリオシド(シアル酸を含むスフィンゴ糖脂質)の役割はいくつも報告されている.たとえば,抗CD3+抗CD28抗体によるTCR刺激に伴って惹起されるガングリオシドGM1の極性化はCD4+ T細胞では起こるが,CD8+ T細胞では起こらない45).さらに,遊走するヒト活性化T細胞では先端部とその対極側とでそれぞれGM1とGM3に富んだ異なるラフトが形成される46).しかし,コレラ毒素Bサブユニット(CTx-B)はラフトの可視化や機能解析に長らく汎用されているが,これで識別されるラフトはその一部であり(後述),構成脂質分子の違いに基づく多様なラフトの存在が考えられる.加えて,細胞の種類やその分化に依存してGM1発現量そのものに大きな違いがあることや,細胞に発現するガングリオシド分子種としてGM1はマイナー成分であるケースも多く知られている.
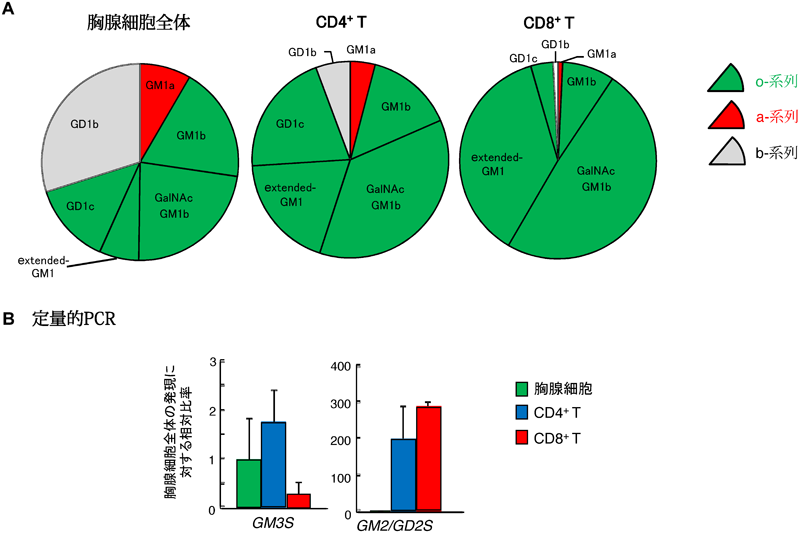
CD4+ T細胞およびCD8+ T細胞にどのようなガングリオシドが発現しているのかをLC-MS/MSで解析したところ,T細胞に発現するガングリオシドはT細胞サブセットごとに特徴的であり,CD4+ T細胞はGM1などのa-系列(図1B参照)の発現が維持されているが,CD8 T細胞はGD1alphaやGalGalNAc-GM1b(extended GM1b)などのo-系列(図1B参照)優位に変化していた(図9A)47,48).さらに,このガングリオシド発現パターンの相違と一致して,GM3Sの遺伝子発現はCD4+ T細胞の方がCD8+ T細胞より約3倍高く,反対にGM2/GD2Sの発現はCD8+ T細胞の方が高い傾向を示した(図9B)47).すなわち,ガングリオシド合成経路のLacCer分岐において,CD4+ T細胞ではGM3Sの活性によってa-系列に合成が進みやすく,CD8+ T細胞ではCD4+ T細胞と比してo-系列に合成が進みやすいことが考えられる.T細胞サブセットにおけるガングリオシド発現パターンの相違が免疫機能に影響するか否かを解析するために,ガングリオシド生合成に関与する二つの鍵酵素であるGM3SおよびGM2/GD2Sの各遺伝子改変マウス(KO)を用いた.GM3S KOはすべてのa-およびb-系列のガングリオシドが欠損してo-系列のみ発現しており,GM2/GD2S KOはGM3とGD3を除くすべてのガングリオシドが欠損している(図1B参照).
当研究室の永福正和博士は,各KOマウスにおけるT細胞の分化および成熟を検討したところ,胸腺,脾臓,リンパ節におけるリンパ球数は野生型マウスと同程度であった.また,胸腺におけるCD4+/CD8+T細胞の割合にも変化はなく,リンパ節および脾臓におけるT細胞/B細胞比およびCD4+ T細胞/CD8+ T細胞比も野生型マウスと同様の分布を示した.次に,野生型および各KOマウスより単離したCD4+ T細胞およびCD8+ T細胞を,抗CD3抗体および抗CD28抗体にて刺激しTCR依存性の反応を検討したところ,GM3S KOではCD4+ T細胞の増殖応答およびサイトカイン産生が重度に低下していたがCD8+ T細胞ではまったく正常であった.対照的に,GM2/GD2S KOでは,CD4+ T細胞の増殖応答およびサイトカイン産生はまったく正常であったがCD8+ T細胞では重度に低下していた47).GM3S KOのCD4+ T細胞およびGM2/GD2S KOのCD8+ T細胞に対してアデノウイルスを介してそれぞれGM3SおよびGM2/GD2Sを遺伝子導入したところ,両者ともに活性化は正常レベルに回復した.さらに,各種ガングリオシド(o-系列のGA2,GA1,GM1b,GD1alpha,a-系列のGM3,GM1,GD1a,b-系列のGD3,GD1b,GT1bおよびLacCer)を外来性に補充しながら,KOマウスT細胞のTCR依存性反応を検討したところ,GM3S KOのCD4+ T細胞ではGM3,GM1あるいはGD1a(すなわちa-系列)を添加したときのみ活性化が正常に回復し,GM2/GD2S KOのCD8+ T細胞では,GA2,GA1,GM1bあるいはGD1alpha(すなわちo-系列)を添加したときのみ活性化が正常に回復した(表2)47).以上のように,CD4+ T細胞はGM3Sによって合成されたa-系列が,CD8+ T細胞ではGM2/GD2Sにより合成されたo-系列がTCR媒介性の活性化に重要であることが判明した.
表2 CD4 T細胞とCD8 T細胞の活性化には,それぞれ特定のガングリオシド分子種を必要とする47,53) | CD4+ T細胞の活性化 | CD8+ T細胞の活性化 |
|---|
| GM3S | GM2/GD2S | GM3S | GM2/GD2S |
|---|
| null | null | null | null |
|---|
| None | ↓↓↓ | Normal | Normal | ↓↓↓ |
| a-系列 | GM3 | Restored | Normal | Normal | ↓↓↓ |
| GM1a | Restored | Normal | Normal | ↓↓↓ |
| GM1b | Restored | Normal | Normal | ↓↓↓ |
| b-系列 | GD3 | ↓↓↓ | Normal | Normal | ↓↓↓ |
| GD1b | ↓↓↓ | Normal | Normal | ↓↓↓ |
| GT1b | ↓↓↓ | Normal | Normal | ↓↓↓ |
| c-系列 | GA2 | ↓↓↓ | Normal | Normal | Restored |
| GA1 | ↓↓↓ | Normal | Normal | Restored |
| GM1b | ↓↓↓ | Normal | Normal | Restored |
| LacCer | ↓↓↓ | Normal | Normal | ↓↓↓ |
| CD4+ T細胞はGM3Sによって合成されたa-系列が,CD8+ T細胞ではGM2/GD2Sにより合成されたo-系列がTCR媒介性の活性化に重要である. |
近年,Th1とTh2に加えて,Th17および制御性T細胞(Treg)が新たなCD4+ T細胞サブセットとして認知されてきた.アレルギー性気道炎症では,Th2(促進的)とTreg(抑制的)のバランスがTh2側にシフトしている48).Th17はまだ未解明な部分も多いが,刺激に伴い炎症性サイトカインであるIL-17を強く産生する細胞であり,好中球性ステロイド抵抗性の重度の喘息に関与し,Th2媒介性の気道炎症も増強するといわれている49).GM3S欠損マウスではTh17へのin vitroの分化誘導が抑制されているという報告がされた50).対照的に,GM3欠損マウスではコラーゲン誘導性関節炎(関節リウマチのマウスモデル)の進行が促進しており,免疫後の局所リンパ節におけるIL-17産生細胞の誘導が亢進するという報告もある51).CD4+ T細胞はGD1c陽性IL-2産生Th1様細胞とGD1c陰性IL-4産生Th2様細胞とに分けることができる11).以上より,CD4+ T細胞サブセットごとに発現するガングリオシド分子種が違い,それに基づく多様なラフトが存在し,アレルギー疾患や自己免疫疾患に影響を与えていることが強く示唆される.今後の研究が期待される.
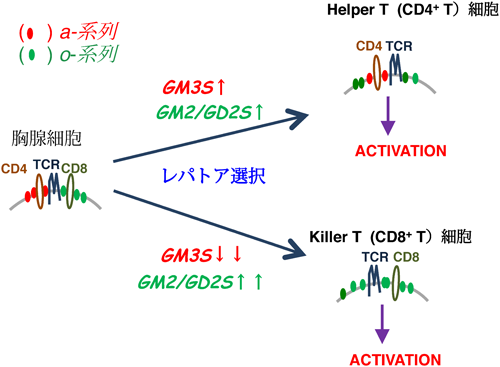
これらの知見から我々は,胸腺におけるT細胞分化過程(レパトア選択)は,T細胞サブセットごとの糖脂質発現の選択でもあり,この糖脂質選択は機能的T細胞への成熟に不可欠な現象であること,すなわち,“Glycolipid selection is indispensable for functional repertoire selection”という作業仮説を提唱している(図10)47,52–54).
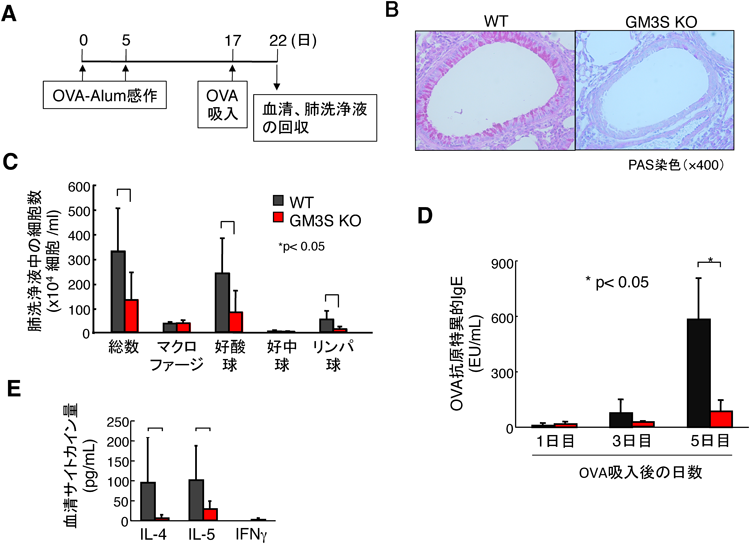
GM3S依存性のガングリオシド分子種a-系列特異的なCD4 T細胞の機能発現が示されたことから,CD4 T細胞免疫応答を基本病態とする気管支喘息におけるガングリオシドの役割を喘息モデルマウスにて本学病態生理学研究室の大野勲教授と検討した.卵白アルブミン(OVA)にて感作後,OVA吸入により,肺洗浄液中のTh2サイトカインと好酸球,リンパ球の増加,すなわちアレルギー性気道炎症が惹起される(図11A).野生型マウスと比較してGM3S KOでは,組織学的にも気道分泌の抑制が確認され(図11B),好酸球,リンパ球(図11C),血清中抗原特異的IgEの出現抑制(図11D)およびTh2サイトカインの著しい減少(図11E)が認められた47).さらに,OVA感作したGM3S欠損マウスのCD4+ T細胞を野生型マウスに移植した実験では炎症細胞の浸潤などが抑制されることから,GM3S欠損マウスにおける気道炎症の抑制はCD4+ T細胞が原因であると実証された47).一方,同様の実験をGM2/GD2S欠損マウスにて行った結果では,WTと有意な違いは認められないことから,in vivoにおけるCD4+ T細胞の免疫機能はGM3S欠損マウスで特異的に抑制される.以上より,少なくともGM3S依存性のガングリオシド分子種はアレルギー性気道炎症の成立に重要な役割を担っていることが判明した.GM3Sおよびその上流にあるグルコシルセラミド合成酵素やラクトシルセラミド合成酵素の阻害がIgE受容体刺激による肥満細胞の活性化経路や脱顆粒を抑制することにより,アレルギー性気道反応を低下させることも報告されている55).現在,さまざまな免疫抑制剤が使用されているが,問題点として非特異的な免疫抑制に基づく重篤な副作用がある.また,T細胞を標的とした免疫抑制剤も試されているが,あらゆるT細胞免疫を抑制することは生体にとって不利益も非常に多く,十分な成果をあげているものはいまだ存在しない.したがって,我々の目指す“ガングリオシド発現制御によるT細胞サブセットの選択的制御”は,T細胞が重要な役割を果たすアレルギー疾患,感染症,自己免疫疾患,腫瘍や移植拒絶反応などに対し,より効果的・安全な治療法を提供することが期待される.
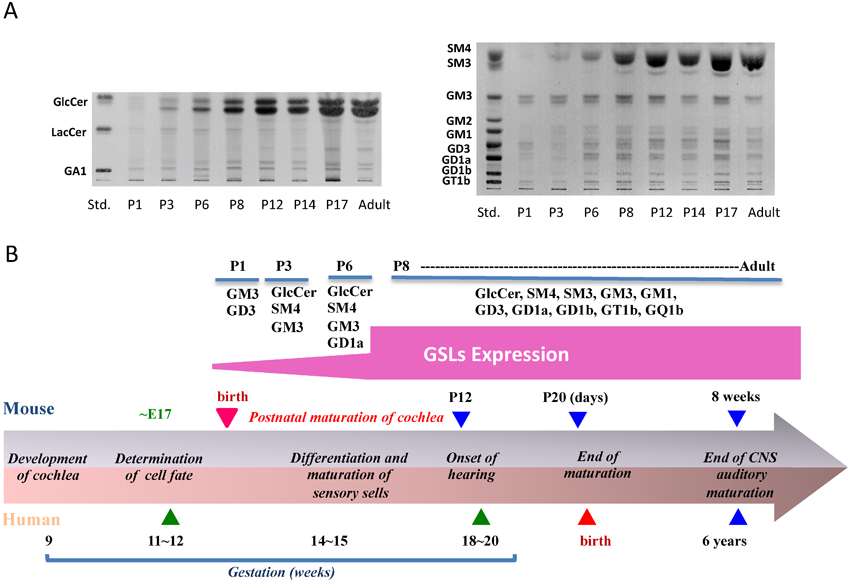
マウスの神経系の酸性糖脂質はヒトを含む他の多くの哺乳類とよく類似しており,硫酸化糖脂質サルファチド[SM4(SO3-GalCer)],ガングリオシドGM1,GD1a,GD1b,GT1b,GQ1などを発現している.脳の形成・発達過程において,酸性糖脂質の発現分子種・発現量は劇的に変化し,生後0日(PO)までにガングリオシドの発現は成熟した脳とほぼ同じ分子種が発現する56).一方,内耳では生後の聴覚機能の成熟期間に対応して,その発現量が劇的に増加し,マウスの脳と内耳には独立したガングリオシド発現制御機構が存在することが判明した(図12)57).ガングリオシドを完全に欠損したマウスであるGM3SとGM2合成酵素(GM2S)の二重欠損(GM2S/GM3S DKO)マウスは,生後すぐに重篤な中枢神経障害を示し,大多数が早期に死亡する58).一方,GM3S KOマウスはメンデルの法則に従う比率で産まれ,ほぼ正常に成長し,寿命も野生型と比較して大きな差はみられない.GM3S KOマウスのほぼすべての臓器では,a-,b-系列のガングリオシドが消失し,通常ほとんど発現がみられないo-系列のガングリオシドが発現する(図1B).前述のようにGM2S/GM3S DKOマウスは生後数か月以内に大半が死亡することから,GM3S KOマウスでは「o-系列」ガングリオシドが生命維持に重要な「a-,b-系列」ガングリオシドの機能を代償していると考えられる.
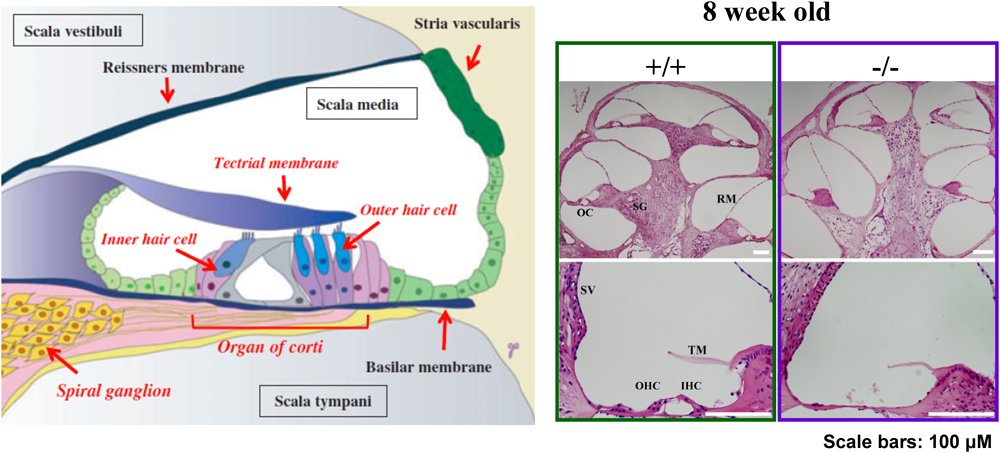
当時大学院生であった吉川弥里さんは,GM3S KOマウスの行動薬理学的検討の目的で,オスとメスのGM3Sヘテロ欠損マウスから生まれる同腹子の遺伝子型判定を行っていた.彼女が「私を振り向いてくれないのがホモ個体です」と言うので,空気圧と音に対する驚愕反応を行ったところ,音に対する反応だけがまったく認められないことが判明し,電気生理学的な聴覚試験(聴性脳幹反応:ABR)においても完全に聴覚機能が障害されていることが確認された59).GM3 KOマウスの聴覚障害の時期は他の難聴モデルマウスと比較しても非常に早く,マウスの聴覚発生時期である生後約14日目ですでに聴力が低下しており,その後数日で完全に消失した.形態学的には,GM3S KOマウスはコルチ器の有毛細胞の選択的な変性・脱落が起こり,遺伝性加齢性難聴モデルや薬剤性難聴と類似した病理所見を示した59)(図13).内耳前庭にも有毛細胞が存在するが,GM3S KOマウスの前庭は組織学的および平衡感覚にも異常は認められなかった.GM3のみを発現するB4galnt1(GM2/GD2S)とSt8sia1(GD3S)の二重欠損マウスでは,このような聴覚の消失は認められていないことから60),GM3はコルチ器の形態・機能維持において重要な役割を有することが考えられた.GM3S KOマウスの結果を合わせて考えると,聴覚機能の維持には,特に“GM3”が重要であると考えられる.この作業仮説を支持する事実として,1)野生型マウスの外有毛細胞の聴毛にはGM3が特異的に発現し,下流のGM1ガングリオシドなどは認められないこと,2)GM3S KOマウスで最初に認められる異常は外有毛細胞の聴毛の変性・脱落であることを見いだしている57).
2009年に我々が発表したGM3S KOマウスの論文には解決しなければならない大きな問題があった59).それはヒトのGM3S欠損症との関連である.2004年,アーミッシュの家系にGM3S欠損の症例が見いだされた61).これはヒトにおけるスフィンゴ糖脂質生合成系酵素の欠損の存在を示した最初の報告である.患者は幼児期に発症する重篤なてんかん症状と視覚異常を示し,20歳前後で死亡する.視覚の異常に関しては網膜の機能異常ではなく,視神経–中枢神経の機能異常によるものであると考えられている62).現在のところ,我々が作製したGM3S KOマウスにおいて,顕著な視覚異常,通常飼育下における重篤なてんかん症状などはみられていない.このヒトとマウスの表現型の違いを解明するには,今後,ガングリオシド組成,遺伝子発現等を多角的に比較検討していく必要がある.このNature Geneticsに掲載された論文には,聴覚機能についての記載はなかった61).しかし,2012年には,フランスで見いだされたGM3S欠損患者で難聴が報告されたことから,GM3S欠損による難聴がヒトでも判明した62).その後,筆者はペンシルバニア州ランカスターのアーミッシュの人々で運営される病院であるClinic for Special Children(CSC)を訪れ,GM3S欠損患者と直に会って,ガングリオシド補給などの治療の可能性を探っている57).
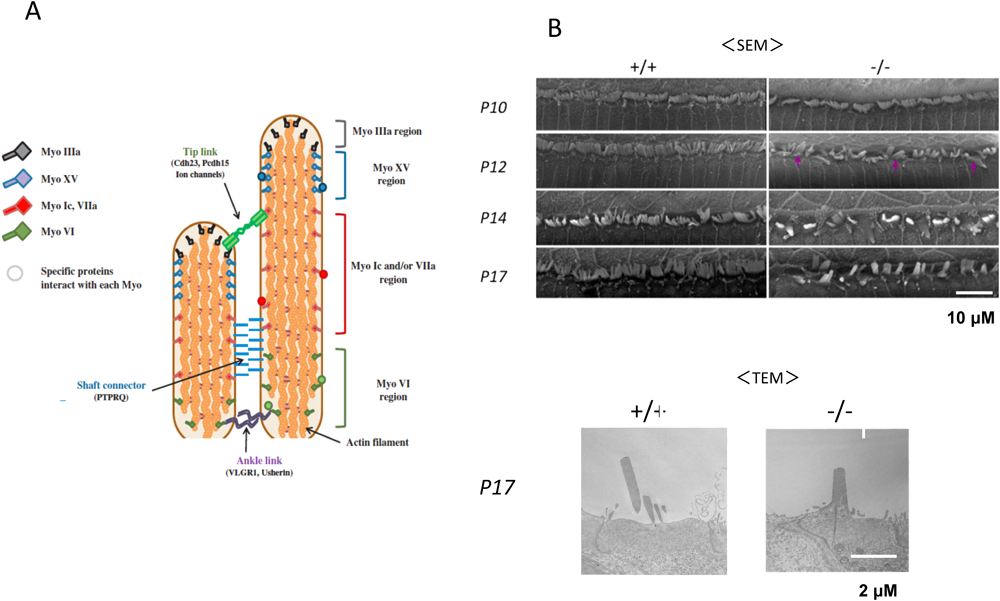
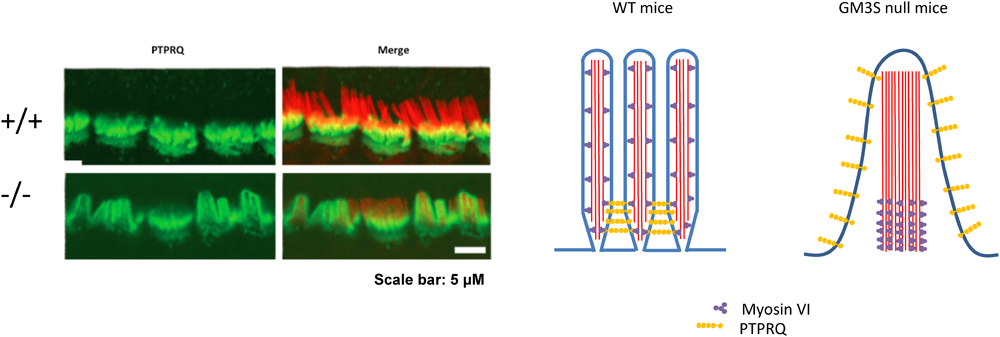
内有毛細胞および外有毛細胞の細胞膜上およびその細胞質内では,各難聴関連タンパク質がそれぞれ特徴的な局在を示し聴毛(不動毛:stereocilia)の形態・機能を維持している(図14A).この聴毛の機能的・形態的維持には同一有毛細胞の聴毛間での分子連結が必須である.当研究室の郷慎司博士と前述の吉川は、コルチ器の形態を詳細に解析したところ,GM3S KOマウスでは,生後まもなく内有毛細胞および外有毛細胞の聴毛の変性が確認された.特に,内有毛細胞では聴毛間の融合,聴毛内の分子の「局在の変化」がGM3S KOマウスの聴覚消失時期に一致して認められた(図14B).この聴毛間の分子連結の崩壊に基づく聴毛の融合がGM3S KOマウスの聴覚機能消失の原因であると考えられる.GM3S KOマウスでは,この連結を担う分子(Shaft connector)の一つPIP2(phosphatidylinositol bisphosphate)の分解をつかさどるPTPRQ(protein tyrosin phosphatase receptor Q)の局在変化を確認している(図15).この他にも,厳密な局在を示すmyosin VI,myosin VIIa,radixinなどの局在が異常を示したことから,細胞膜構成成分ガングリオシドは,有毛細胞の機能性膜領域の形成・維持には必須の成分であることが強く示唆される57).
ガングリオシドは細胞膜上で特定の分子群と相互作用し,マイクロドメイン(機能性微小膜領域)」を形成し,さまざまな生体機能を制御していると考えられている.GM3S KOマウスで変性が顕著にみられた聴毛には,多数の難聴原因遺伝子産物が固有のパターンで発現しており,機能的に異なる複数の機能性領域が存在していることが示唆されている(図14A).したがって,聴毛に特異的に発現する分子の発現,局在,機能をGM3が制御していると考えられる.この有毛細胞における各難聴原因遺伝子産物の選択的輸送システム・局在の分子機構はほとんど解明されていない.ガングリオシドに注目したマイクロドメイン研究の対象としてきわめて興味深い.
感音性難聴は難聴原因遺伝子の変異,加齢や薬物,ストレス,騒音などさまざまな要因によって引き起こされる.先天性の難聴は出生1000人あたりに1人の割合であり,最も頻度が高い先天性疾患の一つである.また,加齢性難聴は65歳以上の40%以上に生じるとされている.さらに,現代社会特有のストレスによる難聴や,携帯音楽プレイヤー等の普及に伴う騒音性の難聴も増加してきている.しかしながら,いったん障害を受け変性した蝸牛内の細胞は再生不可能であることから,現医療においては根本的な感音性難聴の治療はないといってよい.各難聴関連遺伝子の機能はそれぞれ解明されつつある.これら難聴原因遺伝子が中心となって形成される聴覚の機能分子複合体(マイクロドメイン)における糖脂質の役割を解明することから,新たな聴覚機能制御の概念が生まれ,新たな難聴の治療法につながっていくものと期待される.
ガングリオシドを含むスフィンゴ糖脂質分子群の細胞特異的・選択的発現の違いが,インスリンシグナルの恒常性維持,T細胞サブセットの分化・成熟,そして聴覚機能の獲得・維持に必須な生命現象であることが理解されたと思う.今後,自然免疫と獲得免疫による生体恒常性維持機構におけるガングリオシドファミリーの分子種特異的な作動原理を解明し,生理活性脂質としての位置づけを明確にすることが,使命であると考えている.
謝辞Acknowledgments
ミシガン大での博士研究員(フルブライト研究員)時代から始まったガングリオシド研究を今日まで発展できたことは,本文にお名前を述べさせていただいた多くの諸先輩のおかげであり,私の研究人生のさまざまな局面においての出会いに感謝の意を表します.また,最近10年間研究成果の多くは,研究代表者として行ったCREST研究領域:糖鎖の病態機能の解明と利用技術「2型糖尿病の新規な診断・治療技術の開発」(研究統括:谷口直之先生)の期間(2006年から2011年)に見いだされ,現在に至るまで進展させてきたものである.ここに,北海道大学時代そして現在の職場での多くの共同研究者に深謝いたします.
引用文献References
1) 山川民夫(1981)糖脂質物語(講談社学術文庫),講談社.
2) Inokuchi, J. & Radin, N.S. (1987) J. Lipid Res., 28, 565–571.
3) Ishii, A., Ohta, M., Watanabe, Y., Matsuda, K., Ishiyama, K., Sakoe, K., Nakamura, M., Inokuchi, J., Sanai, Y., & Saito, M. (1998) J. Biol. Chem., 273, 31652–31655.
4) Jennermann, R. & Gröne, H.-J. (2013) Prog. Lipid Res., 52, 231–248.
5) Inokuchi, J. (2011) Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci., 87, 179–198.
6) Radin, N.S. & Inokuchi, J. (1988) Biochem. Pharmacol., 37, 2879–2886.
7) Radin, N.S. & Inokuchi, J. (1991) Trends Glycosci. Glycotechnol., 3, 200–213.
8) Radin, N.S., Shayman, J.A., & Inokuchi, J. (1993) Adv. Lipid Res., 26, 183–213.
9) 井ノ口仁一(1998)蛋白質核酸酵素,43, 2495–2502.
10) Lukina, E., Watman, N., Arreguin, E.A., Dragosky, M., Iastrebner, M., Rosenbaum, H., Phillips, M., Pastores, G.M., Kamath, R.S., Rosenthal, D.I., Kaper, M., Singh, T., Puga, A.C., & Peterschmitt, M.J. (2010) Blood, 116, 893–899.
11) Poole, R.M. (2014) Drugs, 74, 1829–1836.
12) Zhao, H., Przybylska, M., Wu, I.-H., Zhang, J., Siegel, C., Komarnitsky, S., Yew, N.S., & Chen, S.H. (2007) Diabetes, 56, 1210–1218.
13) Natoli, T.A., Smith, L.A., Rogers, K.A., Wang, B., Komarnitsky, S., Budman, Y., Belenky, A., Bukanov, N.O., Dackowski, W.R., Husson, H., Russo, R.J., Shayman, J.A., Ledbetter, S.R., Leonard, J.P., & Ibraghimov-Beskrovnaya, O. (2010) Nat. Med., 16, 788–792.
14) Zhao, H., Przybylska, M., Wu, I.H., Zhang, J., Maniatis, P., Pacheco, J., Piepenhagen, P., Copeland, D., Arbeeny, C., Shayman, J.A., Aerts, J.M., Jiang, C., Cheng, S.H., & Yew, N.S. (2009) Hepatology, 50, 85–93.
15) Wennekes, T., van den Berg, R.J.B.H.N., Boot, R.G., van der Marel, G.A., Overkleeft, H.S., & Aerts, J.M.F.G. (2009) Angew. Chem. Int. Ed., 48, 8848–8869.
16) Inokuchi, J., Momosaki, K., Shimeno, H., Nagamatsu, A., & Radin, N.S. (1989) J. Cell. Physiol., 141, 573–583.
17) Inokuchi, J., Usuki, S., & Jimbo, M. (1995) J. Biochem., 117, 766–773.
18) Usuki, S., Hamanoue, M., Kosaka, S., & Inokuchi, J. (1996) J. Neurochem., 67, 1821–1830.
19) Igarashi, K., Furuse, H., Fujii, S., Ito, K., Kaneko, K., Kato, H., Waki, H., Ando, S., & Inokuchi, J. (2002) Glycobiology, 12, 339–344.
20) Yamagishi, K., Ohgami, Y., Iwasaki, K., Jimbo, M., Masuda, H., Fujiwara, M., Igarashi, Y., & Inokuchi, J. (2003) Eur. J. Pharmacol., 462, 53–60.
21) Inokuchi, J. (2009) Int. Rev. Neurobiol., 85, 319–336.
22) Inokuchi, J., Jimbo, M., Kumamoto, Y., Momosaki, K., Shimeno, H., & Nagamatsu, A. (1993) Clin. Exp. Metastasis, 11, 27–36.
23) Hanada, K., Kumagai, K., Yasuda, S., Miura, Y., Kawano, M., Fukasawa, M., & Nishijima, M. (2003) Nature, 426, 803–809.
24) D’Angelo, G., Polishchuk, E., Di Tullio, G., Santoro, M., Di Campli, A., Godi, A., West, G., Bielawski, J., Chuang, C.C., van der Spoel, A.C., Platt, F.M., Hannun, Y.A., Polishchuk, R., Mattjus, P., & De Matteis, M.A. (2007) Nature, 449, 62–67.
25) D’Angelo, G., Uemura, T., Chuang, C.C., Polishchuk, E., Santoro, M., Ohvo-Rekilä, H., Sato, T., Di Tullio, G., Varriale, A., D’Auria, S., Daniele, T., Capuani, F., Johannes, L., Mattjus, P., Monti, M., Pucci, P., Williams, R.L., Burke, J.E., Platt, F.M., Harada, A., & De Matteis, M.A. (2013) Nature, 501, 116–120.
26) Giraudo, C.G. & Maccioni, H.J. (2003) J. Biol. Chem., 278, 40262–40271.
27) Uemura, S., Yoshida, S., Shishido, F., & Inokuchi, J. (2009) Mol. Biol. Cell, 20, 3088–3100.
28) Uemura, S., Shishido, F., Kashimura, M., & Inokuchi, J. (2015) Glycobiology, in press
29) Uemura, S., Kurose, T., Suzuki, T., Yoshida, S., Ito, M., Saito, M., Igarashi, Y., & Inokuchi, J. (2006) Glycobiology, 16, 258–270.
30) Hotamisligil, G.S., Schargill, N.S., & Spiegelman, B.M. (1993) Science, 259, 87–91.
31) Tagami, S., Inokuchi, J., Kabayama, S., Yoshimura, H., Kitamura, F., Uemura, S., Ogawa, C., Ishii, A., Saito, M., Ohtsuka, Y., Sakaue, S., & Igarashi, Y. (2002) J. Biol. Chem., 277, 3085–3092.
32) Nagafuku, M., Sato, T., Sato, S., Shimizu, K., Taira, T., & Inokuchi, J. (2015) Glycobiology, 25, 303–318.
33) Kabayama, K., Sato, T., Saito, K., Loberto, N., Prinetti, A., Sonnino, S., Kinjo, M., Igarashi, Y., & Inokuchi, J. (2007) Proc. Natl. Acad. Sci. USA, 104, 13678–13683.
34) Inokuchi, J. (2007) Yakugaku Zasshi, 127, 579–586.
35) Inokuchi, J. (2010) FEBS Lett., 584, 1864–1871.
36) Inokuchi, J. (2014) Glycoconj. J., 31, 193–197.
37) 井ノ口仁一(2013)実験医学,31, 1566–1573.
38) Feve, B. (2005) Best Pract. Res. Clin. Endocrinol. Metab., 19, 483–499.
39) Inokuchi, J. (2015) Sugar Chains (Suzuki, T., et al., eds.), pp. 219–234, Springer.
40) Grundy, S.M. (2006) Nat. Rev. Drug Discov., 5, 295–309.
41) Senn, H.J., Orth, M., Fitzke, E., Wieland, H., & Gerok, W. (1989) Eur. J. Biochem., 181, 657–662.
42) Sato, T., Nihei, Y., Nagafuku, M., Tagami, S., Chin, R., Kawamura, M., Miyazaki, S., Suzuki, M., Sugawara, S., Takahashi, Y., Saito, A., Igarashi, Y., & Inokuchi, J. (2008) Obes. Res. Clin. Pract., 2, 231–238.
43) Veillon, L., Go, S., Matsuyama, W., Suzuki, A., Nagasaki, M., Yatomi, Y., & Inokuchi, J. (2015) PLoS ONE, 10.1371/journal.pone.0129645
44) Fooksman, D.R., Vardhana, S., Vasiliver-Shamis, G., Liese, J., Blair, D.A., Waite, J., Sacristán, C., Victora, G.D., Zanin-Zhorov, A., & Dustin, M.L. (2010) Annu. Rev. Immunol., 28, 79–105.
45) Kovacs, B., Maus, M.V., Riley, J.L., Derimanov, G.S., Koretzky, G.A., June, C.H., & Finkel, T.H. (2002) Proc. Natl. Acad. Sci. USA, 99, 15006–15011.
46) Gómez-Móuton, C., Abad, J.L., Mira, E., Lacalle, R.A., Gallardo, E., Jiménez-Baranda, S., Illa, I., Bernad, A., Mañes, S., & Martínez-A, C. (2001) Proc. Natl. Acad. Sci. USA, 98, 9642–9647.
47) Nagafuku, M., Okuyama, K., Onimaru, Y., Suzuki, A., Odagiri, Y., Yamashita, T., Iwasaki, K., Fujiwara, M., Takayanagi, M., Ohno, I., & Inokuchi, J. (2012) Proc. Natl. Acad. Sci. USA, 109, E336–E342.
48) Shalaby, K.H. & Martin, J.G. (2010) Curr. Opin. Pharmacol., 10, 218–225.
49) Lloyd, C.M. & Hessel, E.M. (2010) Nat. Rev. Immunol., 10, 838–848.
50) Zhu, Y., Gumlaw, N., Karman, J., Zhao, H., Zhang, J., Jiang, J.L., Maniatis, P., Edling, A., Chuang, W.L., Siegel, C., Shayman, J.A., Kaplan, J., Jiang, C., & Cheng, S.H. (2011) J. Biol. Chem., 286, 14787–14794.
51) Tsukuda, Y., Iwasaki, N., Seito, N., Kanayama, M., Fujitani, N., Shinohara, Y., Kasahara, Y., Onodera, T., Suzuki, K., Asano, T., Minami, A., & Yamashita, T. (2012) PLoS ONE, 7, e40136.
52) Inokuchi, J., Nagafuku, M., Ohno, I., & Suzuki, A. (2013) Cell. Mol. Life Sci., 70, 3067–3075.
53) Nagafuku, M. & Inokuchi, J. (2013) Trends Glycosci. Glycotechnol., 25, 159–169.
54) Inokuchi, J., Nagafuku, M., Ohno, I., & Suzuki, A. (2015) Biochim. Biophys. Acta, 1851, 98–106.
55) Karman, J., Tedstone, J.L., Nathan, K., Gumlaw, N.K., Zhu, Y., Nelson Yew, N., Craig Siegel, C., Shuling Guo, S., Siwkowski, A., Ruzek, M., Jiang, C., & Cheng, S.H. (2010) Int. Immunol., 22, 593–603.
56) Xu, Y.-H., Barnes, S., Ying Sun, Y., & Grabowski, G.A. (2010) J. Lipid Res., 51, 1643–1675.
57) Yoshikawa, M., Go, S., Suzuki, S., Suzuki, A., Katori, Y., Morlet, T., Gottlieb, SM., Fujiwara, M., Iwasaki, K., Strauss, K., & Inokuchi, J. (2015) Hum. Mol. Genet., 24, 2796–2807.
58) Yamashita, T., Wu, Y.P., Sandhoff, R., Werth, N., Mizukami, H., Ellis, J.M., Dupree, J.L., Geyer, R., Sandhoff, K., & Proia, R.L. (2005) Proc. Natl. Acad. Sci. USA, 102, 2725–2730.
59) Yoshikawa, M., Go, S., Takasaki, K., Kakazu, Y., Ohashi, M., Nagafuku, M., Kabayama, K., Sekimoto, J., Suzuki, S., Takaiwa, K., Kimitsuki, T., Matsumoto, N., Komune, S., Kamei, D., Saito, M., Fujiwara, M., Iwasaki, K., & Inokuchi, J. (2009) Proc. Natl. Acad. Sci. USA, 106, 9483–9488.
60) Kawai, H., Allende, M.L., Wada, R., Kono, M., Sango, K., Deng, C., Miyakawa, T., Crawley, J.N., Werth, N., Bierfreund, U., Sandhoff, K., & Proia, R.L. (2001) J. Biol. Chem., 276, 6885–6888.
61) Simpson, M.A., Cross, H., Proukakis, C., Priestman, D.A., Neville, D.C., Reinkensmeier, G., Wang, H., Wiznitzer, M., Gurtz, K., Verganelaki, A., Pryde, A., Patton, M.A., Dwek, R.A., Butters, T.D., Platt, F.M., & Crosby, A.H. (2004) Nat. Genet., 36, 1225–1229.
62) Farukhi, F., Dakkouri, C., Wang, H., Wiztnitzer, M., & Traboulsi, E.I. (2006) Ophthalmic Genet., 27, 89–91.
63) Abe, A., Radin, N.S., Shayman, J.A., Wotring, L.L., Zipkin, R.E., Sivakumar, R., Ruggieri, J.M., Carson, K.G., & Ganem, B. (1995) J. Lipid Res., 36, 611–621.
64) Abe, A., Gregory, S., Lee, L., Killen, P.D., Brady, R.O., Kulkarni, A., & Shayman, J.A. (2000) J. Clin. Invest., 105, 1563–1571.
65) Jimbo, M., Yamagishi, K., Yamaki, T., Nunomura, K., Kabayama, K., Igarashi, Y., & Inokuchi, J. (2000) J. Biochem., 127, 485–491.
著者紹介Author Profile
 井ノ口 仁一(いのくち じんいち)
井ノ口 仁一(いのくち じんいち)東北薬科大学分子生体膜研究所所長.機能病態分子学教室教授.博士(薬学).
略歴福岡県に生まれる.福岡大学薬学部を卒業.同大学院,生化学教室助手を経て,1985年ミシガン大学医学部神経化学研究所博士研究員.1991年生化学工業東京研究所主任研究員を経て,1998年北海道大学大学院薬学研究科助教授.2006年より現職.
研究テーマと抱負自然免疫と獲得免疫による生体恒常性維持機構におけるスフィンゴ糖脂質の分子種特異的な作動原理を解明し,生理活性脂質としての位置付けを明確にすることが,使命であると考えている.
趣味音楽鑑賞,ガーデニング.