1) Thomson, J.A., Itskovitz-Eldor, J., Shapiro, S.S., Waknitz, M.A., Swiergiel, J.J., Marshall, V.S., & Jones, J.M. (1998) Science, 282, 1145–1147.
2) Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., & Yamanaka, S. (2007) Cell, 131, 861–872.
5) Wells, J.M. & Melton, D.A. (2000) Development, 127, 1563–1572.
6) Kanai-Azuma, M., Kanai, Y., Gad, J.M., Tajima, Y., Taya, C., Kurohmaru, M., Sanai, Y., Yonekawa, H., Yazaki, K., Tam, P.P., & Hayashi, Y. (2002) Development, 129, 2367–2379.
7) Monaghan, A.P., Kaestner, K.H., Grau, E., & Schutz, G. (1993) Development, 119, 567–578.
8) Yasunaga, M., Tada, S., Torikai-Nishikawa, S., Nakano, Y., Okada, M., Jakt, L.M., Nishikawa, S., Chiba, T., Era, T., & Nishikawa, S. (2005) Nat. Biotechnol., 23, 1542–1550.
9) Iwashita, H., Shiraki, N., Sakano, D., Ikegami, T., Shiga, M., Kume, K., & Kume, S. (2013) PLoS ONE, 8, e64291.
10) Brown, S., Teo, A., Pauklin, S., Hannan, N., Cho, C.H., Lim, B., Vardy, L., Dunn, N.R., Trotter, M., Pedersen, R., & Vallier, L. (2011) Stem Cells, 29, 1176–1185.
11) Teo, A.K., Arnold, S.J., Trotter, M.W., Brown, S., Ang, L.T., Chng, Z., Robertson, E.J., Dunn, N.R., & Vallier, L. (2011) Genes Dev., 25, 238–250.
12) Bertero, A., Madrigal, P., Galli, A., Hubner, N.C., Moreno, I., Burks, D., Brown, S., Pedersen, R.A., Gaffney, D., Mendjan, S., Pauklin, S., & Vallier, L. (2015) Genes Dev., 29, 702–717.
13) Loh, K.M., Ang, L.T., Zhang, J., Kumar, V., Ang, J., Auyeong, J.Q., Lee, K.L., Choo, S.H., Lim, C.Y., Nichane, M., Tan, J., Noghabi, M.S., Azzola, L., Ng, E.S., Durruthy-Durruthy, J., Sebastiano, V., Poellinger, L., Elefanty, A.G., Stanley, E.G., Chen, Q., Prabhakar, S., Weissman, I.L., & Lim, B. (2014) Cell Stem Cell, 14, 237–252.
16) Shiraki, N., Shiraki, Y., Tsuyama, T., Obata, F., Miura, M., Nagae, G., Aburatani, H., Kume, K., Endo, F., & Kume, S. (2014) Cell Metab., 19, 780–794.
17) Chetty, S., Pagliuca, F.W., Honore, C., Kweudjeu, A., Rezania, A., & Melton, D.A. (2013) Nat. Methods, 10, 553–556.
18) Herrera, P.L. (2000) Development, 127, 2317–2322.
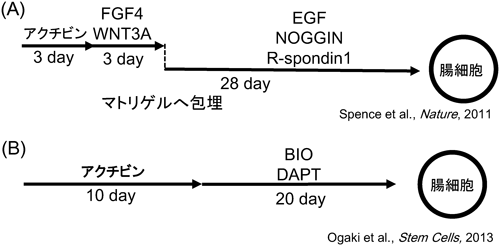
19) Ogaki, S., Harada, S., Shiraki, N., Kume, K., & Kume, S. (2011) BMC Dev. Biol., 11, 13.
21) Shapiro, A.M., Ricordi, C., Hering, B.J., Auchincloss, H., Lindblad, R., Robertson, R.P., Secchi, A., Brendel, M.D., Berney, T., Brennan, D.C., Cagliero, E., Alejandro, R., Ryan, E.A., DiMercurio, B., Morel, P., Polonsky, K.S., Reems, J.A., Bretzel, R.G., Bertuzzi, F., Froud, T., Kandaswamy, R., Sutherland, D.E., Eisenbarth, G., Segal, M., Preiksaitis, J., Korbutt, G.S., Barton, F.B., Viviano, L., Seyfert-Margolis, V., Bluestone, J., & Lakey, J.R. (2006) N. Engl. J. Med., 355, 1318–1330.
22) D’Amour, K.A., Bang, A.G., Eliazer, S., Kelly, O.G., Agulnick, A.D., Smart, N.G., Moorman, M.A., Kroon, E., Carpenter, M.K., & Baetge, E.E. (2006) Nat. Biotechnol., 24, 1392–1401.
23) Kroon, E., Martinson, L.A., Kadoya, K., Bang, A.G., Kelly, O.G., Eliazer, S., Young, H., Richardson, M., Smart, N.G., Cunningham, J., Agulnick, A.D., D’Amour, K.A., Carpenter, M.K., & Baetge, E.E. (2008) Nat. Biotechnol., 26, 443–452.
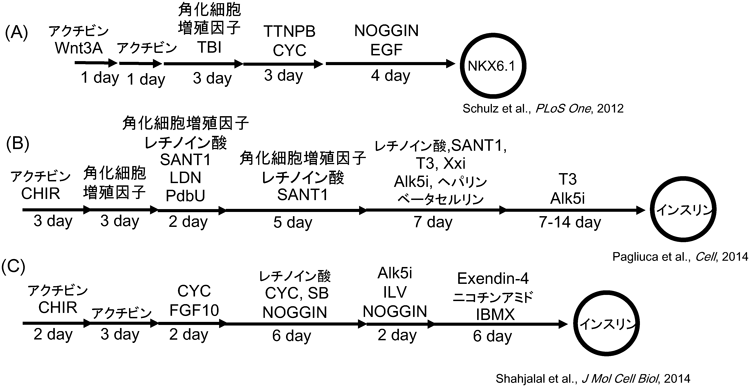
24) Schulz, T.C., Young, H.Y., Agulnick, A.D., Babin, M.J., Baetge, E.E., Bang, A.G., Bhoumik, A., Cepa, I., Cesario, R.M., Haakmeester, C., Kadoya, K., Kelly, J.R., Kerr, J., Martinson, L.A., McLean, A.B., Moorman, M.A., Payne, J.K., Richardson, M., Ross, K.G., Sherrer, E.S., Song, X., Wilson, A.Z., Brandon, E.P., Green, C.E., Kroon, E.J., Kelly, O.G., D’Amour, K.A., & Robins, A.J. (2012) PLoS ONE, 7, e37004.
25) Sasada, R., Ono, Y., Taniyama, Y., Shing, Y., Folkman, J., & Igarashi, K. (1993) Biochem. Biophys. Res. Commun., 190, 1173–1179.
26) Pagliuca, F.W., Millman, J.R., Gurtler, M., Segel, M., Van Dervort, A., Ryu, J.H., Peterson, Q.P., Greiner, D., & Melton, D.A. (2014) Cell, 159, 428–439.
27) Fu, A., Ng, A.C., Depatie, C., Wijesekara, N., He, Y., Wang, G.S., Bardeesy, N., Scott, F.W., Touyz, R.M., Wheeler, M.B., & Screaton, R.A. (2009) Cell Metab., 10, 285–295.
28) Granot, Z., Swisa, A., Magenheim, J., Stolovich-Rain, M., Fujimoto, W., Manduchi, E., Miki, T., Lennerz, J.K., Stoeckert, C.J. Jr., Meyuhas, O., Seino, S., Permutt, M.A., Piwnica-Worms, H., Bardeesy, N., & Dor, Y. (2009) Cell Metab., 10, 296–308.
29) Rezania, A., Bruin, J.E., Arora, P., Rubin, A., Batushansky, I., Asadi, A., O’Dwyer, S., Quiskamp, N., Mojibian, M., Albrecht, T., Yang, Y.H., Johnson, J.D., & Kieffer, T.J. (2014) Nat. Biotechnol., 32, 1121–1133.
30) Shahjalal, H.M., Shiraki, N., Sakano, D., Kikawa, K., Ogaki, S., Baba, H., Kume, K., & Kume, S. (2014) J. Mol. Cell Biol., 6, 394–408.
31) Sakano, D., Shiraki, N., Kikawa, K., Yamazoe, T., Kataoka, M., Umeda, K., Araki, K., Mao, D., Matsumoto, S., Nakagata, N., Andersson, O., Stainier, D., Endo, F., Kume, K., Uesugi, M., & Kume, S. (2014) Nat. Chem. Biol., 10, 141–148.
33) Si-Tayeb, K., Noto, F.K., Nagaoka, M., Li, J., Battle, M.A., Duris, C., North, P.E., Dalton, S., & Duncan, S.A. (2010) Hepatology, 51, 297–305.
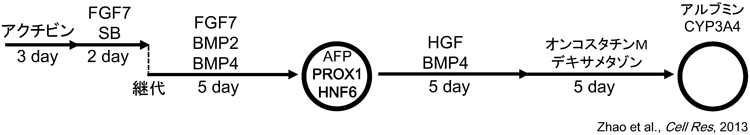
34) Zhao, D., Chen, S., Duo, S., Xiang, C., Jia, J., Guo, M., Lai, W., Lu, S., & Deng, H. (2013) Cell Res., 23, 157–161.
35) Seth, A., Ye, J., Yu, N., Guez, F., Bedford, D.C., Neale, G.A., Cordi, S., Brindle, P.K., Lemaigre, F.P., Kaestner, K.H., & Sosa-Pineda, B. (2014) Development, 141, 538–547.
36) Umeda, K., Suzuki, K., Yamazoe, T., Shiraki, N., Higuchi, Y., Tokieda, K., Kume, K., Mitani, K., & Kume, S. (2013) Stem Cell Res. (Amst.), 10, 179–194.
37) Takebe, T., Sekine, K., Enomura, M., Koike, H., Kimura, M., Ogaeri, T., Zhang, R.R., Ueno, Y., Zheng, Y.W., Koike, N., Aoyama, S., Adachi, Y., & Taniguchi, H. (2013) Nature, 499, 481–484.
39) Barker, N., van Es, J.H., Kuipers, J., Kujala, P., van den Born, M., Cozijnsen, M., Haegebarth, A., Korving, J., Begthel, H., Peters, P.J., & Clevers, H. (2007) Nature, 449, 1003–1007.
40) Walton, K.D., Kolterud, A., Czerwinski, M.J., Bell, M.J., Prakash, A., Kushwaha, J., Grosse, A.S., Schnell, S., & Gumucio, D.L. (2012) Proc. Natl. Acad. Sci. USA, 109, 15817–15822.
42) Sato, T., Vries, R.G., Snippert, H.J., van de Wetering, M., Barker, N., Stange, D.E., van Es, J.H., Abo, A., Kujala, P., Peters, P.J., & Clevers, H. (2009) Nature, 459, 262–265.
43) Spence, J.R., Mayhew, C.N., Rankin, S.A., Kuhar, M.F., Vallance, J.E., Tolle, K., Hoskins, E.E., Kalinichenko, V.V., Wells, S.I., Zorn, A.M., Shroyer, N.F., & Wells, J.M. (2011) Nature, 470, 105–109.
44) Watson, C.L., Mahe, M.M., Munera, J., Howell, J.C., Sundaram, N., Poling, H.M., Schweitzer, J.I., Vallance, J.E., Mayhew, C.N., Sun, Y., Grabowski, G., Finkbeiner, S.R., Spence, J.R., Shroyer, N.F., Wells, J.M., & Helmrath, M.A. (2014) Nat. Med., 20, 1310–1314.
45) Shiraki, N., Umeda, K., Sakashita, N., Takeya, M., Kume, K., & Kume, S. (2008) Genes Cells, 13, 731–746.
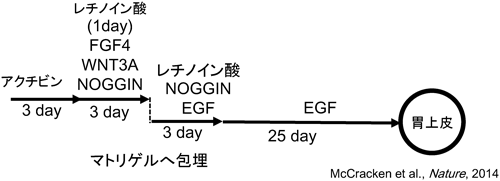
48) McCracken, K.W., Cata, E.M., Crawford, C.M., Sinagoga, K.L., Schumacher, M., Rockich, B.E., Tsai, Y.H., Mayhew, C.N., Spence, J.R., Zavros, Y., & Wells, J.M. (2014) Nature, 516, 400–404.
50) Munshi, L., Keshavjee, S., & Cypel, M. Lancet. (2013) Respir. Med., 1, 318–328.
52) Hogan, B.L., Barkauskas, C.E., Chapman, H.A., Epstein, J.A., Jain, R., Hsia, C.C., Niklason, L., Calle, E., Le, A., Randell, S.H., Rock, J., Snitow, M., Krummel, M., Stripp, B.R., Vu, T., White, E.S., Whitsett, J.A., & Morrisey, E.E. (2014) Cell Stem Cell, 15, 123–138.
53) Green, M.D., Chen, A., Nostro, M.C., d’Souza, S.L., Schaniel, C., Lemischka, I.R., Gouon-Evans, V., Keller, G., & Snoeck, H.W. (2011) Nat. Biotechnol., 29, 267–272.
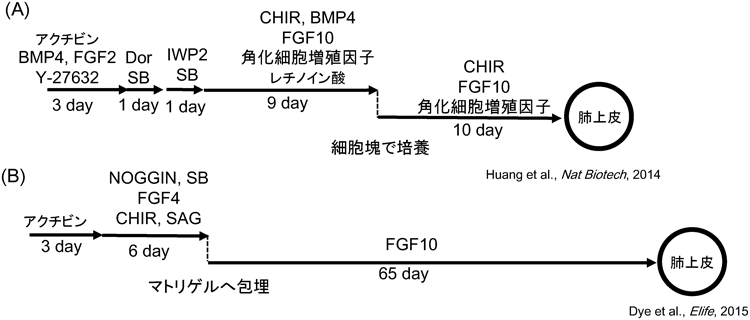
54) Huang, S.X., Islam, M.N., O'Neill, J., Hu, Z., Yang, Y.G., Chen, Y.W., Mumau, M., Green, M.D., Vunjak-Novakovic, G., Bhattacharya, J., & Snoeck, H.W. (2014) Nat. Biotechnol., 32, 84–91.
55) Dye, B.R., Hill, D.R., Ferguson, M.A., Tsai, Y.H., Nagy, M.S., Dyal, R., Wells, J.M., Mayhew, C.N., Nattiv, R., Klein, O.D., White, E.S., Deutsch, G.H., & Spence, J.R. (2015) eLife, 4.
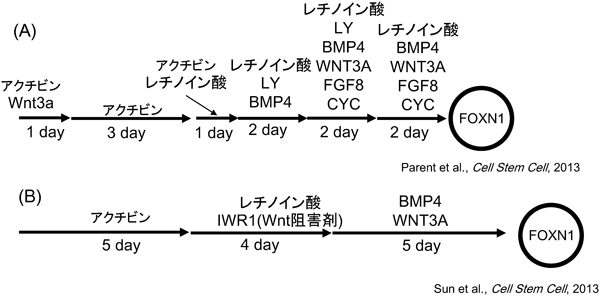
58) Parent, A.V., Russ, H.A., Khan, I.S., LaFlam, T.N., Metzger, T.C., Anderson, M.S., & Hebrok, M. (2013) Cell Stem Cell, 13, 219–229.
59) Sun, X., Xu, J., Lu, H., Liu, W., Miao, Z., Sui, X., Liu, H., Su, L., Du, W., He, Q., Chen, F., Shi, Y., & Deng, H. (2013) Cell Stem Cell, 13, 230–236.
60) Soh, C.L., Giudice, A., Jenny, R.A., Elliott, D.A., Hatzistavrou, T., Micallef, S.J., Kianizad, K., Seach, N., Zuniga-Pflucker, J.C., Chidgey, A.P., Trounson, A., Nilsson, S.K., Haylock, D.N., Boyd, R.L., Elefanty, A.G., & Stanley, E.G. (2014) Stem Cell Rev., 2, 925–937.