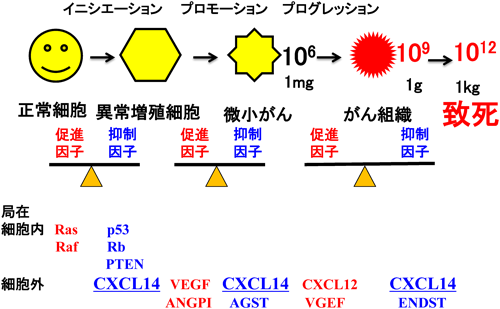
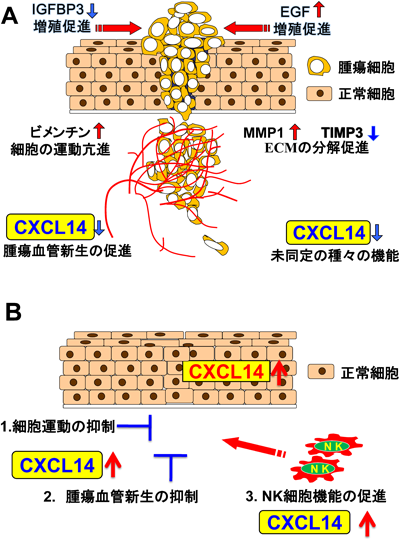
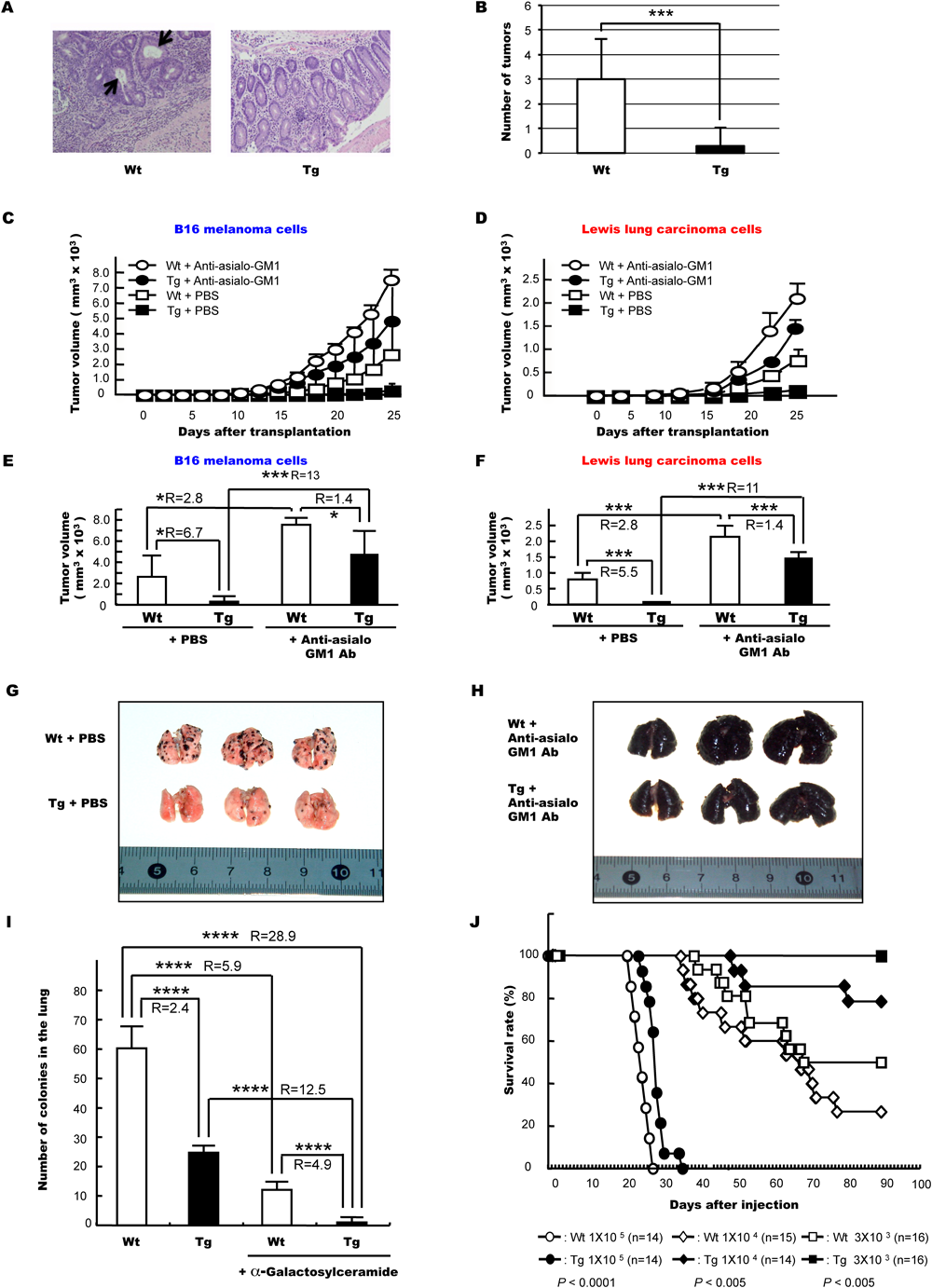
がんに強いマウスを作る:がんの分子標的予防医学の開発に向けてProduction and characterization of cancer resistant mouse: Toward development of molecular preventive medicine of cancer
1 神奈川歯科大学大学院歯学研究科Graduate School of Dentistry, Kanagawa Dental University ◇ 〒238-8580 神奈川県横須賀市稲岡町82番地Inaoka-cho 82, Yokosuka-shi, Kanagawa 238-8580, Japan
2 神奈川歯科大学口腔難治疾患研究センターOral Health Science Research Center, Kanagawa Dental University ◇ 〒238-8580 神奈川県横須賀市稲岡町82番地Inaoka-cho 82, Yokosuka-shi, Kanagawa 238-8580, Japan
受付日:2015年5月7日Received: May 7, 2015
発行日:2015年10月25日Published: October 25, 2015