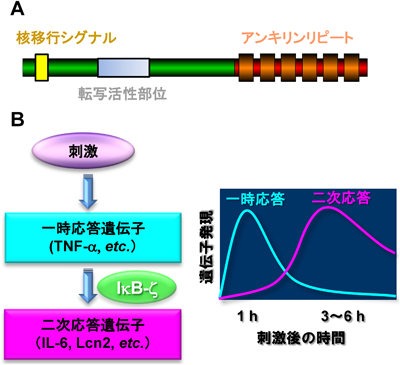
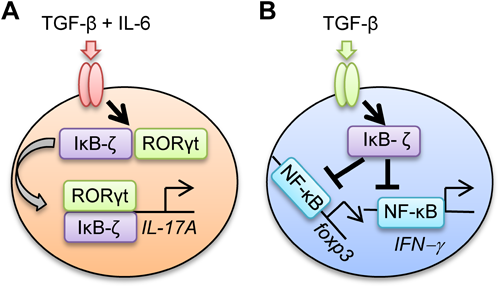
核内IκB-ζによる炎症応答の制御Nuclear IκB-ζ controls inflammatory responses
1 岐阜大学大学院医学系研究科Graduate School of Medicine, Gifu University ◇ 〒501-1194 岐阜県岐阜市柳戸1番1号Yanagito 1-1, Gifu-shi, Gifu 501-1194, Japan
2 東北大学大学院生命科学研究科Graduate School of Life Sciences, Tohoku University ◇ 〒980-8577 宮城県仙台市青葉区片平二丁目1番1号Katahira 2-1-1, Aoba-ku, Sendai-shi, Miyagi 980-8577, Japan
受付日:2015年7月10日Received: July 10, 2015
発行日:2015年10月25日Published: October 25, 2015