Rap1を介したリンパ球移動の制御機構Regulation of lymphocyte trafficking
北里大学理学部School of Science, Kitasato University ◇ 〒252-0373 神奈川県相模原市南区北里一丁目15番1号Kitazato 1-15-1, Minami-ku, Sagamihara-shi, Kanagawa 252-0373, Japan
発行日:2015年12月25日Published: December 25, 2015
免疫システムは活発な免疫細胞の生体内移動を基盤としており,時間的・空間的に厳密に制御されることによって秩序が維持され,生体防御機能を発揮できる.リンパ球は血流を介して全身を移動しているが,リンパ節へ到達すると高内皮細静脈(HEV: high endothelial venule)からリンパ節内に移動(ホーミング)し,特異抗原を探索する.こうしたリンパ球のリンパ節巡回は獲得免疫の開始に必須である.インテグリンを介する接着・移動は,このリンパ球のホーミングや抗原探索の基盤となっている.低分子量Gタンパク質Rap1は,下流標的分子として,2種のタンパク質(RAPLとMst1)を活性化することで,インテグリンを介する細胞移動を促進している.今回,Mst1の下流でRab13が,この過程に重要な役割を果たしていることを突き止めたので紹介する.
© 2015 公益社団法人日本生化学会© 2015 The Japanese Biochemical Society
我々は生活環境中に存在する無数の微生物に取り囲まれているが,そう簡単には感染症を発症することがない.免疫システムが病原体などの異物を監視し,侵入を速やかに察知し撃退しているからである.その際に重要なのは,外敵がいつどこから侵入しても速やかに対応することを可能にしている,リンパ球の血流を介する全身移動とその制御である.すなわち,免疫システムが常に全身を監視できるのは,血流に乗って移動しているリンパ球が,必要なときに適切な部位の血管内皮細胞に接着し,そこを通り抜けて目的部位に到達できるからである.
骨髄や胸腺などの中枢リンパ組織(あるいは一次リンパ組織)で産生されたリンパ球は,血管系を介してリンパ節や脾臓などの二次リンパ組織へ移動するが,そのリンパ球固有の抗原受容体が認識する特異抗原に出会わない限りは,輸出リンパ管から出て,リンパ液中を移動し,左鎖骨下静脈へ続く胸管から血液中に戻る.そして再びリンパ節へ移動するという現象を恒常的に繰り返している.このリンパ球再循環現象(lymphocyte recirculation)1)は,一度も特異抗原に出会っていないリンパ球が特異抗原に出会うために重要である.なぜなら,直接,外敵の侵入を見張っているのは,外界と接する皮膚や粘膜に存在する樹状細胞と呼ばれる細胞で,感染が起こるとその情報を獲得免疫系の司令塔であるTリンパ球に伝えるため,リンパ節へ移動するからである2).すなわち,樹状細胞は,通常は皮膚の表皮細胞間や腸管粘膜固有層などに高い貪食能を有する未熟樹状細胞として存在する.外敵が侵入しそれを捉えると,劇的に変化し,それまでの組織内での拘束を逃れ,リンパ管を通ってリンパ節へ移動し,高い抗原提示能を有する成熟樹状細胞へと分化し,病原体を抗原へ加工し,Tリンパ球へ提示する3).一方,血流を介して移動しているリンパ球は,リンパ節内に存在する特殊な血管である高内皮細静脈(HEV)に到達すると停止し,通り抜けてリンパ節内に入る.リンパ節に入った後も,リンパ節内の場を構成するストローマ細胞(細綱細胞)のネットワーク上を活発に遊走しながら,自分の抗原受容体が認識する特異抗原を提示する樹状細胞を走査する4).Tリンパ球は,特異抗原を提示する樹状細胞に出会うと停止し,両者は強く結合することで,抗原受容体を介して活性化され,獲得免疫が開始する.
リンパ球は,血管内皮細胞上でセレクチン・インテグリンという接着分子を介する多段階の連続した接着カスケードを経て,血管の外に移動することがわかっている5).リンパ球の細胞表面上に発現するL-セレクチンは,HEV上の細胞表面に発現するシアリルルイス6-スルホXと呼ばれる硫酸化糖鎖を認識する.血管内で両者の結合はリンパ球の減速(ローリング)を起こすが,この現象は,ホーミングの第1段階として重要であることがわかっている6).ローリングしているリンパ球は,この間に血管内皮細胞上のヘパラン硫酸プロテオグリカンと結合し提示されたケモカインの刺激を受け,細胞表面上に発現するインテグリンが活性化される.これによりリンパ球はHEV上で停止し,リンパ節内に入ることができる.また,インテグリンは,リンパ球が特異抗原を探してリンパ節内のストローマ細胞ネットワーク上を遊走し,樹状細胞上を走査する際の足場として,免疫応答の始動に寄与している.さらにリンパ球はインテグリンを介して樹状細胞に強固に接着することで,抗原情報を受け取り活性化し,分化する.
このようにインテグリンの接着活性は,リンパ球が血管内にとどまっているときには「オフ」になっているが,リンパ節のHEVや感染部位の血管内皮細胞上にケモカインが提示されていると「オン」になり,インテグリンを介する接着を足場に血管の外へ移動することができる.こうした仕組みは,リンパ球上に発現するインテグリンの構造および局在が,ケモカイン刺激で速やかにダイナミックに変化することで発揮される.我々は,ケモカインの刺激を受けてインテグリンの活性を上昇させる細胞内シグナル伝達分子を探索し,低分子量Gタンパク質のRap1を同定した7).更に,Rap1の下流の標的分子として,2種のタンパク質(RAPLとMst1)を単離した(図1)8,9).これらの欠損マウスを作製し解析したところ,いずれのマウスのリンパ球もインテグリンを介する接着が低下し,二次リンパ組織へのホーミングは低下していた10,11).一方,このシグナル伝達経路がリンパ球の極性形成を誘導し,先端部にインテグリンを集めることで,接着・遊走を促進することを明らかにした.Mst1はRap1が活性化されRAPLと会合すると,リン酸化酵素活性が上昇し,その局在が核周辺領域から先端部へ秒単位で移動することから,Rap1-RAPL-Mst1シグナルはおそらく小胞輸送を介したインテグリンの局在化を促し,リンパ球の接着や遊走を促進させると考えられた(図1)10).そこで小胞輸送に重要なRabファミリー低分子Gタンパク質の中に,Mst1の下流でインテグリンの極性輸送に関与する分子を検索し,Rab13を同定した.さらに,Mst1とRab13が協調してインテグリンクラスターを含む先端膜(接着膜)を誘導する機構を解明した12).
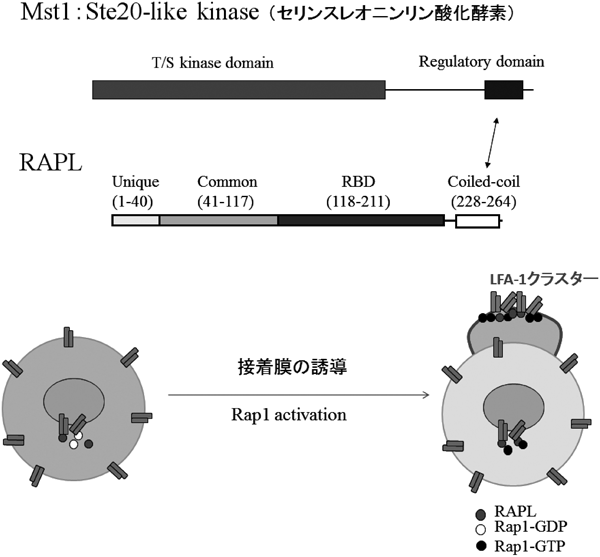
(上)Mst1はste20-like kinaseで,RAPLはC末端側にcoiled-coil domainを持つアダプター分子.RAPLのcoiled-coil domainを介して,Mst1のregulatory domainに会合すると,Mst1のリン酸化酵素活性は上昇する.(下)Rap1活性化によって形成されるLFA-1クラスターは,新生膜(接着膜)形成とともに誘導される.
Rap1優性活性型変異体(Rap1V12)をリンパ球に発現させると,細胞は特徴的な先端部(leading edge)と後端部(uropod)を形成し,極性を持った形態をとる.これとともにリンパ球に発現する主たるインテグリンであるLFA-1は先端部に集積しクラスターを形成し,一方,CD44分子は後端に集積し,細胞表面受容体の分離が起こる(図2).この形態は,リンパ球をケモカインで刺激したときに誘導される形態と同様であった.このLFA-1クラスターがリガンドであるICAM-1への接着上昇に重要なことは,Rap1V12を発現させても接着の上昇が起きないRap1不応答LFA-1変異体(LFA-1はαL鎖とβ2鎖からなるヘテロ二量体で,αL鎖の細胞内領域の1095番目からC末端側を削除した変異体,αL1095)が先端部に局在できないことから明らかになった(図2).このαL1095は先端膜を除く形質膜上には発現することから,先端膜とそれ以外の形質膜は交わっておらず,細胞内からの膜輸送によって形成された新生膜であると考えられた(図1).下流標的分子であるMst1-RAPLはショ糖密度勾配遠心法によって同じ小胞画分に濃縮されること,および免疫電子顕微鏡法で小胞の細胞質側に局在することから,RAPL-Mst1は小胞輸送に関与し,LFA-1を極性輸送し接着膜を誘導しているという仮説が裏づけられた(図3).
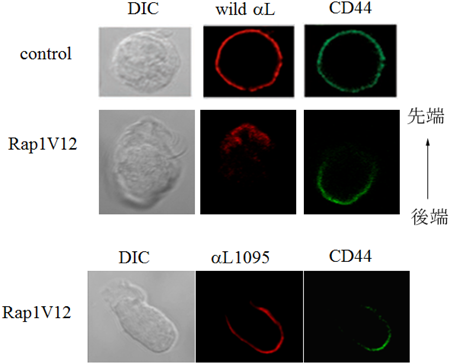
Rap1V12を導入した細胞は,極性を持った形に変化し,LFA-1は先端部に集積し,CD44は後端に局在する(上).一方,LFA-1のαL鎖の細胞内領域の1095番目からC末端側を削った変異体(αL1095)は先端部に存在しない(下).
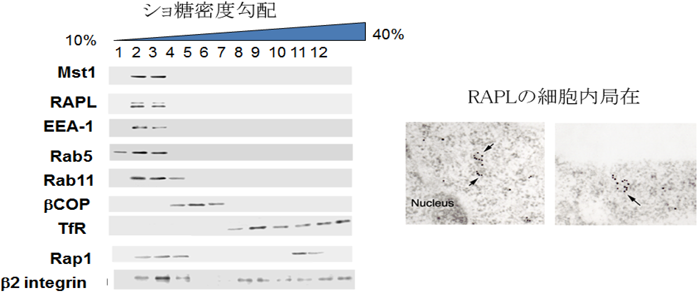
(左)マウスリンパ球を用いて,ショ糖密度勾配遠心法で分画すると,RAPL-Mst1は,EEA-1やRab5と同様の軽い小胞画分に濃縮される.Rap1およびLFA-1(β2インテグリン)は,形質膜画分と小胞画分に存在する.(右)RAPLの局在を金コロイド法で電子顕微鏡を用いて観察した像.小胞の細胞質側に局在する.
そこで小胞輸送に関与するRabファミリーGTPaseについて,Mst1とin vitroで会合し,COS細胞での一過性発現系で共局在を示す分子をスクリーニングした.同定された候補分子について,優性活性型および優性抑制型変異体をBAF/LFA-1細胞(ヒトLFA-1遺伝子を導入したIL-3依存性のproB細胞株)へ過剰発現させた結果,Rab13がICAM-1への接着および移動に関与することが示唆された.
Rab13優性抑制型変異体および優性活性型変異体とMst1の会合を検討した結果,両者とも会合するが,活性型Rab13がより強くMst1に会合することが判明した.また,Rab13はケモカイン刺激によって活性化されることが,既知の下流標的分子であるMICAL-L2タンパク質のRBD(Rab13 binding domain)とGST(glutathione S-transferase)の融合タンパク質を用いたpull-down法で明らかとなった.そこで,Mst1がケモカインによるRab13の活性化に関与する可能性を,Mst1をノックダウンした細胞を用いて検討したところ,Rab13の活性化が低下することが判明した.
Rab13のGDP/GTP交換因子であるDENND1Cに着目し,DENND1CがMst1によってリン酸化されるかどうか,また,それがGEF活性の上昇につながるかどうかを,まず,COS細胞を用いた過剰発現系を用いて調べた.DENND1CはMst1と会合しリン酸化されること,それによってRab13に対するGEF(GDP/GTP exchange factor)活性が上昇することが判明した.さらに,ケモカイン刺激によって,DENND1CがMst1依存性にリン酸化され,DENND1Cをノックダウンした細胞では,ケモカイン刺激によるRab13活性化が低下することがわかった(図4).これらのことから,Mst1はDENND1Cのリン酸化を介してケモカインによるRab13活性化を誘導していることが明らかとなった.
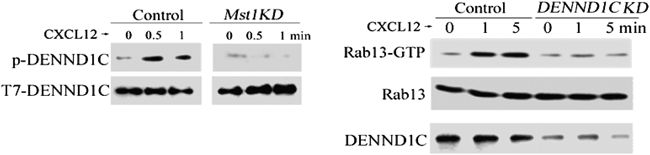
(左)BAF細胞(proB細胞,control細胞)をCXCL12で刺激すると,30秒後にDENND1Cのリン酸化が上昇するが,Mst1をノックダウンした細胞(Mst1KD)では,DENND1Cのリン酸化の上昇は認められなかった.(右)DENND1Cをノックダウンした細胞(DENND1CKD)では,CXCL12刺激によるRab13活性化(Rab13-GTPの増加)が生じなかった.
Rab13優性抑制型変異体を導入,およびノックダウンした細胞では,ケモカインによって誘導されるLFA-1クラスターの形成が生じず,LFA-1-ICAM-1を介する接着および遊走が低下することがわかった(図5).また,Rab13の局在を検討したところ,無刺激では核周辺領域および形質膜に存在するが,ケモカイン刺激後は先端膜のLFA-1クラスター部に集積することを見いだした(図5).Mst1をノックダウンした細胞やRab13優性抑制型変異体(Rab13DN)を導入した細胞では,ケモカイン刺激後のRab13の先端部での集積は認められなかった(図5).これらの結果より,Rab13は,ケモカイン刺激によって誘導されるLFA-1クラスターの形成に必須であること,ならびに,Mst1依存性にケモカインよって活性化されると,Rab13も先端膜へ移行することが明らかとなった.
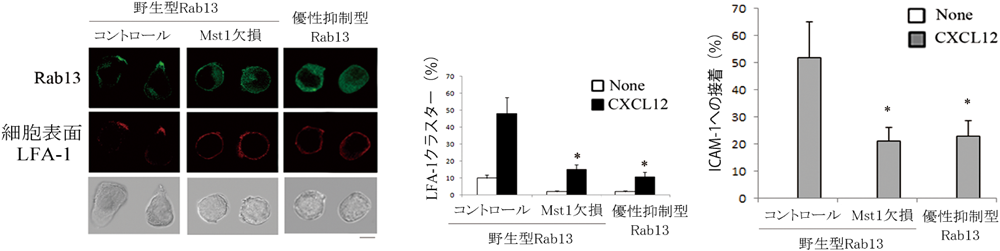
(左)ケモカイン刺激によって形成されるLFA-1クラスター部(赤)に野生型Rab13が集積している.Rab13優性抑制型変異体を発現させた細胞ではLFA-1クラスターの形成が阻害され,優性抑制型Rab13は細胞質に存在していた.(中央)優性抑制型Rab13は,Mst1欠損細胞と同様に,ケモカイン(CXCL12)によるLFA-1クラスター形成が阻害された.(右)優性抑制型Rab13発現細胞ではLFA-1クラスター形成の低下とともに,ICAM-1への接着が低下した.
次に異なる蛍光タンパク質を融合したRab13とLFA-1のケモカイン刺激後の細胞内局在の変化をライブイメージングで観察した.無刺激の状態で,両者の細胞内での局在は核周辺部でほぼ一致しており,ケモカイン刺激後は,両者とも先端部へ同時に移行することがわかった.このことから,Rab13がLFA-1を含む小胞を極性輸送していることが示唆された.
Rap1シグナルによるLFA-1クラスター形成は,αL鎖の細胞内領域にRAPL-Mst1が会合することで生じることが明らかになっているので,Rab13がMst1の下流でLFA-1クラスター形成に関与するとすれば,αL鎖の細胞内領域を削除した変異体(αL1095)はRab13集積部位に存在しないはずである.この仮説を検証するために,αL1095とRab13に異なる蛍光タンパク質を融合させ,BAF細胞に導入し,ケモカイン刺激後のこれらの局在の変化を観察した.ケモカイン刺激前は,Rab13とαL1095変異体も,核周辺部と一様に形質膜に共局在を示すが,刺激後は,Rab13が先端部に移行し集積するのに対し,αL1095はRab13クラスターから排除されていることが確認された8).このことから,Rab13はαL鎖細胞内領域に会合したRAPL-Mst1依存性にLFA-1を前方へ極性輸送していると考えられた.
Rab13およびLFA-1の前方への移動時,しばしば線状のアクチンケーブルあるいは微小管に沿って存在すると思われる像が観察されたので,アクチン繊維に特異的に結合するLifeactおよび微小管の先端部に局在するEB3にRab13と異なる蛍光タンパク質を融合させ,Rab13がケモカイン刺激によって,前方へ移動するときにそれらの局在がどのように変化するかを観察した.Rab13はLifeactに沿って前方へ移動し,Rab13が先端部へ移行した後に,アクチン繊維が脱重合することがわかった(図6).EB3とRab13は局在の一致が認められなかった.このことから,アクチンケーブルに沿ってRab13はLFA-1を先端部に極性輸送していることが示唆された.
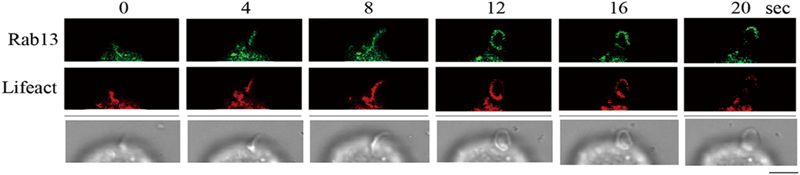
ケモカイン刺激によってRab13の前方へ移動時のアクチン繊維の動態を観察するため,繊維化アクチンに特異的に結合するLifeactにmcherryを融合させたタンパク質を発現させ,ケモカイン刺激後の変化をライブで観察した.Rab13はアクチン繊維に沿って移動し,Rab13の先端部集積後にアクチン繊維は消失した.
アクチン繊維の伸展は,VASP(vasodilator-stimulated phosphoprotein)ファミリー分子が先端部に局在し,束化因子やアクチン重合因子をリクルートすることで生じることが明らかとなっている.VASPの157番目のセリン残基のリン酸化はVASPがアクチン繊維先端部に局在するのに必須であることがわかっている.そこで,Mst1がVASPの157番目のセリンをリン酸化する可能性を,COS細胞を用いた過剰発現系で検討したところ,Mst1の過剰発現によってVASPのリン酸化が上昇することがわかった.さらにVASPがケモカイン刺激で一過性にリン酸化され,Mst1をノックダウンした細胞では,リン酸化のレベルが低下していた.VASPも,Mst1依存性にケモカイン刺激によってリン酸化される基質であることが明らかとなった.
Lifeactとは異なる蛍光タンパク質を融合させたVASPを導入した細胞を用いて,ケモカイン刺激後のアクチンケーブル発達時におけるVASPの局在を検討したところ,VASPがアクチンケーブルの先端部に集積していることがわかった(図7).VASPの157番目のセリンをアラニンに変えた変異体(S157A)は,アクチンケーブルの先端部には局在せず,S157A変異体の過剰発現によってアクチン繊維の伸展は阻害され,LFA-1クラスター形成も低下することが明らかとなった(図7).
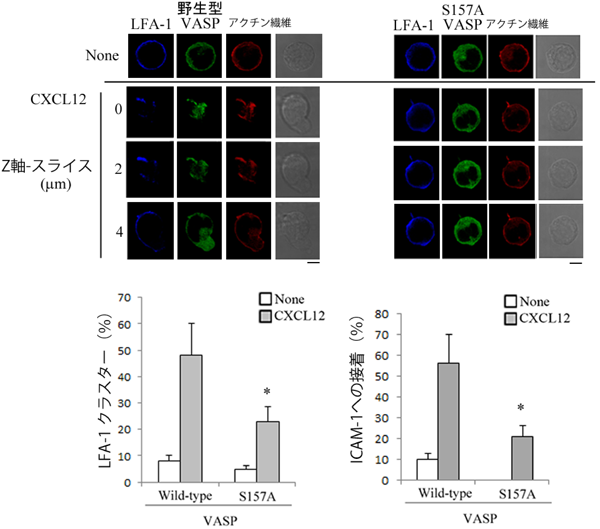
(左)野生型VASPは,伸長しているアクチン繊維の先端部に集積し,LFA-1クラスターが形成されている.VASP S157A変異体は,アクチン繊維の先端部に集積せず,LFA-1クラスターは形成されていない.(中央)CXCL12刺激によって形成されるLFA-1クラスターは,VASP S157A変異体を発現させた細胞株では有意に低下していた.(右)CXCL12刺激によって誘導されるICAM-1への接着も有意に低下した.
Rab13と同じファミリーに属するRab8やRab10は,ミオシンVをリクルートすることで小胞をアクチンケーブル依存性に輸送することが報告されている.そこでミオシンVaとRab13-GTPの結合をin vitroおよび細胞内で調べたところ,ミオシンVaはRab13-GTPとのみ会合することがわかった.次にミオシンVaとRab13を異なる蛍光タンパク質と融合させ導入した細胞を用いて,ケモカイン刺激後の両者の変化をライブイメージングで観察したところ,ケモカイン刺激前は一致していないが,ケモカイン刺激後に先端部でのみ部分的に一致していた.ミオシンVaの優性抑制型変異体の過剰発現はLFA-1クラスター形成およびICAM-1への接着を低下させることが明らかとなった.
これらのことから,Mst1とRab13が協調してLFA-1クラスターを形成するメカニズムについて模式図(図8)にまとめた.すなわち,Rap1活性化によりRAPL-Mst1複合体がLFA-1の細胞内領域に結合する.Mst1はVASPの157番目のセリンをリン酸化することによってアクチンの伸展を促進するとともに,DENNS1Cをリン酸化して,Rab13の活性化が起こる.Rab13-GTPは,Mst1に会合するとともにミオシンVaをリクルートし,アクチンケーブル依存性にLFA-1を先端部に極性輸送している.
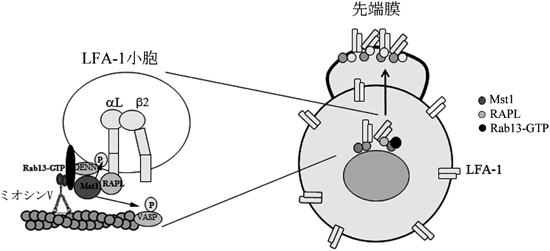
Rap1活性化により,RAPL-Mst1はLFA-1のα鎖の細胞内領域に会合するとともに,Mst1のリン酸化酵素活性が上昇する.Mst1はVASPをリン酸化することでアクチンケーブルの伸展を誘導する.一方,DENND1Cをリン酸化することでRab13に対するGEF(GTP/GDP交換因子)が上昇し,Rab13の活性化が生じる.Rab13-GTPはミオシンVaをリクルートすることで,アクチンケーブルに沿ってLFA-1を前方へ極性輸送する.
Rab13を欠損したマウスを作製し,そのマウスのリンパ組織から回収したリンパ球(Rab13欠損リンパ球)のLFA-1を介する接着・遊走における役割を解析した.その結果,Rab13欠損リンパ球において,ケモカイン刺激によって誘導されるLFA-1クラスターの形成が損なわれ,ICAM-1上での接着・遊走が低下することが判明した(図9).そのため,リンパ球は二次リンパ組織にホーミングできず,Rab13欠損マウスではリンパ球が減少し,二次リンパ組織(脾臓,リンパ節)が低形成となった(図10).このことから,リンパ球動態制御がRab13の生体内における主な役割の一つであることが明らかになった.
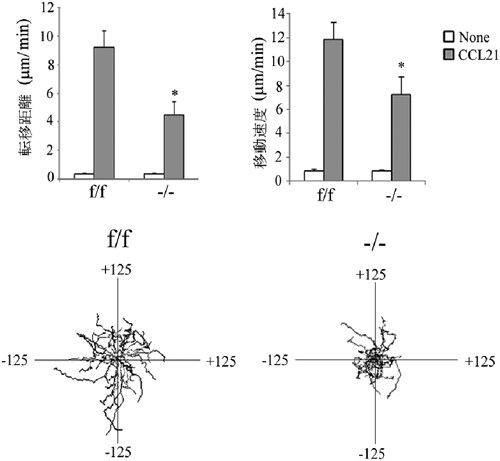
正常(f/f)およびRab13欠損(−/−)T細胞の,精製したマウスICAM-1上での遊走を,ケモカイン(CCL21)存在下および非存在下で,タイムラップス撮影し,metamorph softwareを用いて,転移距離(Displacement)および速度(velocity)を測定した.
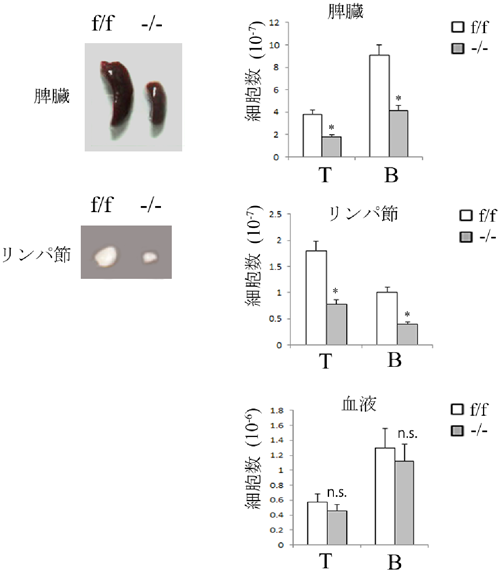
(左)正常(f/f)およびRab13欠損(−/−)マウスの脾臓,リンパ節,(右)f/fおよび−/−マウスの脾臓,リンパ節,血液におけるT細胞数およびB細胞数を示している.
Rap1はHEV上でのLFA-1を介する停止に必須であり,Rap1を欠損すると,ナイーブリンパ球は二次リンパ組織に入ることができなくなる.Rap1の作用のうち,RAPL-Mst1は細胞極性の形成,細胞遊走に関わっており,このメカニズムとしてRab13を介する極性輸送の仕組みを明らかにした.Rap1はLFA-1だけではなく,VLA-4,α4β7を介する接着の上昇も促進する.しかしながら,エフェクター細胞が炎症血管上で停止する際にはRap1を必ずしも必要としないことから,Rap1-GTPは,免疫監視機構に重要なリンパ球再循環現象に専門のインテグリン活性化因子であるといえるかもしれない.
1) Springer, T.A. (1995) Annu. Rev. Physiol., 57, 827–872.
2) Randolph, G.J., Angeli, V., & Swartz, M.A. (2005) Nat. Rev. Immunol., 5, 617–628.
3) Dalod, M., Chelbi, R., Malissen, B., & Lawrence, T. (2014) EMBO J., 33, 1104–1116.
4) Lämmermann, T. & Sixt, M. (2008) Immunol. Rev., 221, 26–43.
5) Butcher, E.C. & Picker, L.J. (1996) Science, 272, 60–66.
6) Sperandio, M., Gleissner, C.A., & Ley, K. (2009) Immunol. Rev., 230, 97–113.
7) Katagiri, K., Hattori, M., Minato, N., Irie, S., Takatsu, K., & Kinashi, T. (2000) Mol. Cell. Biol., 20, 1956–1969.
8) Katagiri, K., Maeda, A., Shimonaka, M., & Kinashi, T. (2003) Nat. Immunol., 4, 741–748.
9) Katagiri, K., Imamura, M., & Kinashi, T. (2006) Nat. Immunol., 7, 919–928.
10) Katagiri, K., Ohnishi, N., Kabashima, K., Iyoda, T., Takeda, N., Shinkai, Y., Inaba, K., & Kinashi, T. (2004) Nat. Immunol., 5, 1045–1051.
11) Katagiri, K., Katakai, T., Ebisuno, Y., Ueda, Y., Okada, T., & Kinashi, T. (2009) EMBO J., 28, 1319–1331.
12) Nishikimi, A., Ishihara, S., Ozawa, M., Etoh, K., Fukuda, M., Kinashi, T., & Katagiri, K. (2014) Sci. Signal., 7, ra72.

北里大学理学部生物科学科教授.獣医学博士.
1981年北海道大学獣医学研究科修士課程卒.83~85年中外製薬新薬研究所研究員.85~88年米国ノースカロライナ大学癌研究所リサーチアソシエイト.94~98年ニッピマトリックス研究所主任研究員.99~2004年京都大学医学研究科バイエル寄付講座分子免疫アレルギー・講師.04~09年関西医科大学分子遺伝学講座准教授.09~12年関西学院大学理工学部生命科学科教授.12年~現職.
研究テーマと抱負リンパ球の動態制御機構.免疫システムは,活発な免疫細胞の生体内移動を基盤としており,時間的・空間的に厳密に制御されています.免疫細胞の生体内移動は,Rap1という低分子量Gタンパク質によって調節されていることを見出し,その作用機序の解明に取り組んでいます.
ウェブサイトhttp://www.kitasato-u.ac.jp/sci/resea/seibutsu/bogyo/seitaibogyo.HP/Home.html
趣味地域猫活動.

This page was created on 2015-10-28T16:08:01.488+09:00
This page was last modified on 2015-12-17T15:42:54.921+09:00
このサイトは(株)国際文献社によって運用されています。