1)mRNA上でのリボソームの滞留による翻訳制御
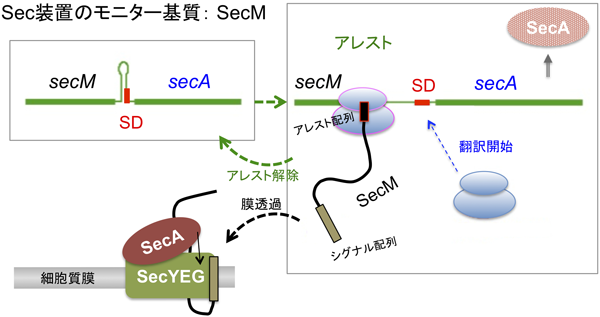
細菌のSec膜透過経路では分泌タンパク質前駆体がSecA ATPaseによって駆動され,SecYEGトランスロコン(ポリペプチド透過孔)を介して膜を越えて輸送される.SecAの発現は,細胞のタンパク質分泌活性の低下に呼応して上昇する.この調節には,secAの上流遺伝子(uORF) secM(170コドン)が関与する15).SecMはN末端にシグナル配列を持つSec経路の基質であるが,ペリプラズムに分泌されても直ちに分解除去されてしまう16).SecM合成途上鎖が膜透過されない状況が起こると,下流遺伝子からのSecAの翻訳が上昇する.SecMの役割は合成途上に発揮され,その効果はcis(同一mRNAからの発現)特異的である.SecMは,自らが基質となってSec膜透過装置の活性をモニターしている.
SecAはsecM-secA mRNAから翻訳される.secAの翻訳開始頻度は,リボソーム結合(SD)配列が利用可能かどうかによって決まる.この遺伝子間領域にはステム–ループ二次構造を形成する反転反復配列があり,その中にsecAのSD配列が含まれるため17),ステム構造が形成されるとSD配列は機能できない.ステム形成配列の5′側隣接領域で伸長アレストが起こると,滞留したリボソームによって二次構造形成が妨げられ,SD配列が遊離リボソームによって利用可能となる(図1).実際,secMの翻訳はこの領域で伸長停止(アレスト)を起こす16).アレストは,SecM合成途上鎖が膜透過を受けることに共役して解除される(図1).正常細胞ではリボソームの滞留は短時間で終わるが,Sec膜透過装置の活性が低下すると,アレスト解除の遅延,リボソーム滞留時間の延長,secA翻訳量の上昇という一連の結果が生ずる.SecMは自らを翻訳するリボソームのmRNA上での滞留時間をSec装置の活性変動に呼応して制御することにより,secA翻訳の基底レベルの維持と分泌欠損に応答した誘導に不可欠の役割を演じている18).したがって,SecMのアレスト機能を損なう変異は致死となる(SecAを過剰に供給すればレスキューされる)18).
2)SecMの合成途上鎖は伸長アレストを起こす
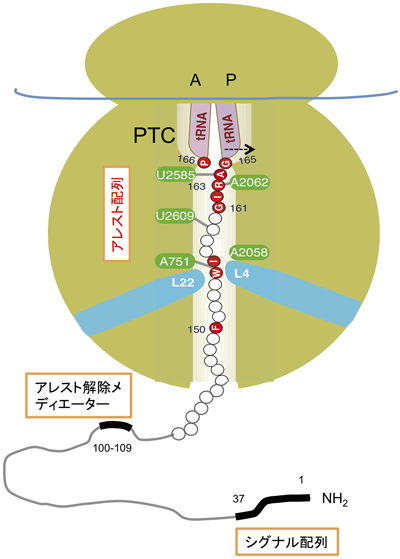
SecMの翻訳はin vivoでもin vitroでも伸長アレストを起こす.in vivoではSec経路を変異導入や薬剤で阻害したときに,アレストが継続する.SecM自体のシグナル配列を欠損させると,Sec装置の活性が正常でも,アレストが継続してsecAの過剰翻訳をもたらす.アレスト解除機構が働かないin vitro翻訳系では,SecMのアレストは緩慢な自発的解除はあるものの安定に継続する.そして,立ち止まったリボソームの位置をトウプリント(toeprint,下流プライマーからの逆転写反応のリボソームによる中断)実験で解析できる.SecMの翻訳はP部位にSecM1-165-tRNAGlyが,A部位にコドン166に対応するtRNAProが存在する状態で停止し,Gly-Proペプチド結合形成が不全で,ピューロマイシンも働けない状態にある(図2)19).アレスト状態はtrans-translation(tRNAとmRNAの機能を併せ持つtmRNAに乗り移って継続される翻訳)やArfA-RF2によるリボソーム救出機構によっても解消できない20,21).異常mRNAでのリボソームの停滞と異なり,遺伝子発現制御の役割を持つ翻訳アレストはむやみに解除されては困るという合目的性に合致している.
SecMの翻訳伸長アレストの原因はアミノ酸配列(アレスト配列)にあり,mRNAの性質ではない16,22).このことは,アレストがC末端付近のフレームシフト変異やアゼチジン(プロリンのアナログ)の取り込みにより損なわれるが,同義変異によって影響を受けないことからわかる.変異解析から同定されたアレストに必要・十分な配列はF150XXXXWIXXXXGIRAGP166(Xは任意のアミノ酸)である.SecMアレスト配列は,無関係タンパク質中に挿入されても翻訳伸長阻止機能を発揮する(例外について,アレスト解除機構の項目で議論する)ため,合成途上鎖—リボソーム複合体を作製する手段として広く利用されている23,24).
3)SecM合成途上鎖はリボソームのPTCやトンネルの成分と相互作用する
PTCで重合したアミノ酸残基は,翻訳伸長の進行に伴い,順次リボソームの脱出トンネルを通って動く.トンネルは,長さ100 Å,内径15 Åほどで,合成途上鎖の最も「若い」部分30~40アミノ酸残基を収容できる.その内壁は主としてRNAで構成されているが,PTCから1/3ほどにある狭窄部位にはリボソームタンパク質L22とL4の一端が顔を出している.細胞のすべてのタンパク質のすべてのアミノ酸残基は,順次位置を変えつつトンネルに収容された状態を経験する.リボソームトンネルが発見された当時,トンネルが合成途上鎖と相互作用することはないと考えられた25).しかし,合成途上鎖のアミノ酸残基とリボソーム構成成分との間に特殊な位置関係が実現するとPTCが機能不全(アレスト)に陥るのであり,翻訳はリボソームと翻訳産物が相互作用する可能性を孕みつつ進行するのである.
新生鎖はトンネル内でαらせん以上の高次構造はとれないとされ26),低温電子顕微鏡解析で観察される合成途上鎖はおおむね伸びた「変性」状態にある27).ただし,Znフィンガー程度の小さな構造はトンネル内で形成可能という報告もなされた28).SecMのアレストに重要な残基は,翻訳アレストを起こしたときPTC付近(G161IRAGP166)から,トンネルの狭窄部位近傍(Ile156,Trp155,Phe150)にかけて位置している(図2).Bernsteinらは,Mannheimia succiniciproducensのSecMアレスト配列ではPTCに近接したH159APIRGSP166(P部位の二つ手前のArg163とA部位のPro166が特に重要)のみが重要であること(図3),アレスト配列には柔軟性があることを報告した29).
トンネルの狭窄部位付近に位置する,23S rRNAのA751やリボソームタンパク質L22のβヘアピン先端の残基91,93,それらの対面壁に位置する23S rRNAのA2058やリボソームタンパク質L4などに変異が起こると,SecMのアレストが損なわれる22).また,PTC近傍の23S rRNA残基A2062,A2503の変異もアレスト欠損を示す30).これらのことから,アレスト配列はリボソームとの特異的相互作用を介して翻訳にブレーキをかけることが示唆される22).低温電子顕微鏡によるSecM翻訳アレスト複合体の構造解析によれば,合成途上鎖はリボソームと複数の部位で接触している31).トンネル内部でのSecM Trp155とrRNA A751,よりN末端側のSecM残基とL22 Lys84など,およびPTCの近傍でのSecM Arg163とrRNA A2062などである.その結果,PTC機能に関わるA2062の配向異常とP部位におけるtRNAGly 3′末端の正常な位置からの約2 Å外側への逸脱が起こる(図2).シミュレーションによればA部位のtRNAProも異常な配向をとる32).Mankinらは,細胞に存在する一般のタンパク質の合成には関わらないため活性中心の変異を得ることができる(オルソゴナルな)リボソームを構築して,アミノ酸収容やPTC反応に関わるA部位のRNA残基A2451がSecMのアレストにも関わることを示した33).合成途上鎖とリボソーム残基間の相互作用の総和として,PTC付近の活性部位残基と基質tRNA双方の構造変化が起こり,翻訳アレストに至るものと思われる.
4)合成途上鎖の動的挙動がアレスト解除に働く
SecM合成途上鎖がSec装置による膜透過反応を受けると伸長アレストが解除される.翻訳過程が産物(新生ポリペプチド鎖)の動的挙動の影響を受けることはセントラルドグマの根本的問題でもある.アレスト解除の機構として,「引っぱり説」と「信号伝達説」が考えられる.前者は合成途上鎖が膜透過に伴って受ける物理力が,後者はSec装置と合成途上鎖の特異的分子認識が,それぞれアレスト解除のきっかけとなるという考えである34).Oliverらは,SecM配列の途中に疎水性の膜貫通領域となる(膜透過を停止させる)配列を挿入するとSecAの翻訳制御が損なわれることをもって,引っぱり説の根拠とした35).Bernsteinらは,引っぱり力が適切なタイミングで,(アレスト配列が翻訳された直後に)合成途上鎖にかかることが,アレスト解除に重要であると提唱した36).SecMシグナル配列の性質およびシグナル配列からアレストモチーフまでの長さが,アレスト解除に適するよう厳密に設計されているという考えである.魅力的な説だが,証明はされていない.
von Heijneらは,疎水性領域(19残基)をSecMの特定の位置に挿入すると翻訳アレストが不全となる(解除される)ことを示した37).挿入位置がPTC(A部位)から数えて30残基上流および40残基上流(アレスト状態におけるトンネルの出口付近)のとき,アレスト解除の効果が最大となった.トンネル出口付近に位置する新生鎖の領域がSecYEGトランスロコンへの挿入および膜脂質層への分配の動きを起こすことによって,物理的力を発生し,アレスト解除に至ると解釈できる.
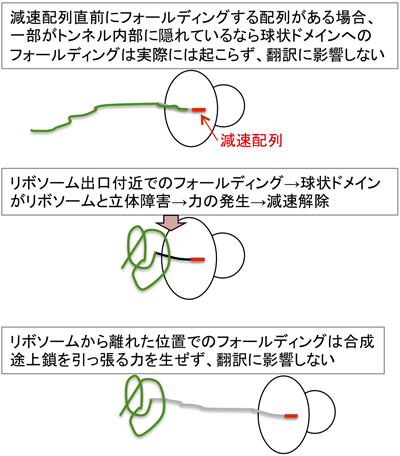
最近,合成途上鎖にかかる物理的力によってアレスト解除が起こることの証明がBustamanteらによる一分子実験によってなされた38).アレスト状態のSecM合成途上鎖—リボソーム複合体を固定し,光ピンセットを用いて合成途上鎖に10 pN程度以上の引っぱり力を加えると,SecMの翻訳が再開され,ピューロマイシンへの転移(固定状態からの破断)として観察された.また,細胞内で合成途上鎖に力を加える手段として,球状ドメインへのフォールディングを考え,フォールディングを起こすTop7配列をSecMと緑色蛍光タンパク質(GEP)の融合タンパク質のSecM部分に挿入した.PTCから32~39残基上流領域域への挿入に限ってアレストが解除されてGFP部分が合成された.翻訳アレストの結果リボソームのPTC部位にtRNAを介して固定されている合成途上鎖のトンネル出口付近に位置する部分が,トンネル内部には入れない大きさの球状ドメインにフォールディングを起こそうとするとき,トンネル出口から十分離れていない場合には,リボソームによる立体的な制約を受けることになる.その結果,リボソームトンネルに収容されたポリペプチド部分に対して引っ張り力を発生することになる.この引っ張り力がアレスト解除の原因となるものと考えられる(図5参照).Top7のフォールディングは12 pN程度の力を生じてアレストを解除させるようだ.さらに小さな構造がトンネル内部で形成された場合も立体障害,力の発生,アレスト解除が起こる28).
アレスト解除の引っぱり説が実証されたとはいえ,野生型SecMの膜透過と共役した翻訳アレスト解除が完全に説明されたわけではない.PTC領域から129残基も上流に位置するシグナル配列に膜透過の力が加わっても,リボソーム内部に波及しないかもしれない.我々は,シグナル配列とアレスト配列によってはさまれたSecMの中央領域の残基100~109(アレスト解除メディエーター領域と命名)の欠失や置換によってアレスト解除の効率が下がることを見いだした39).中央領域はシグナル配列とアレスト配列を結ぶ単なるリンカーではなく,Secモニター機能に重要な役割を担っていることが考えられる(図2).なお,SecMのリボソームの外に出た配列に,「アレスト補強機能」があるという報告40)に関しては,外来配列の影響を考慮した注意深い検討が必要であると考えている.
5)「モニター基質」によるリアルタイムフィードバック制御
通常,フィードバック発現制御では,ある因子の活性の変動を細胞の状態変化や代謝産物の濃度(反応速度ではない)変化によって感知する.SecMによるSecA発現制御では,Sec膜透過装置の活性(反応速度)低下を基質であるSecMが直接リアルタイムに感知する.このような制御因子を「モニター基質」“monitoring substrate”と呼ぶことを提案したい.分泌欠損応答では,Sec因子の変異に呼応したSecAの発現上昇が観察されるが,「自然界でめったに起こらない変異に対する応答機構にどのような生理学的意義があるのか」という質問を受けることがある.SecMによる制御が発動する生理的な条件として低温41)をあげて答えてきた18).しかし,モニター基質による制御は,機能の(微妙な)低下(たとえば基質の過多によっても起こりうる)の結果が表現型として表れないように,リアルタイムで作動できることにこそ意義があるのではないだろうか? 細胞への影響が出る前の根元のところでの「内部フィードバック」とでもいえる制御系なのであり,その効果がSec装置の機能欠損変異を用いてしか示せないとしても,むしろそのこと自体が逆説的に意義深いことであると考えるようになった.
3. さまざまなアレストペプチドからみえてくる翻訳の実像
1)多様なアミノ酸配列が,個別の方式でリボソームと相互作用し,翻訳にブレーキをかけることができる
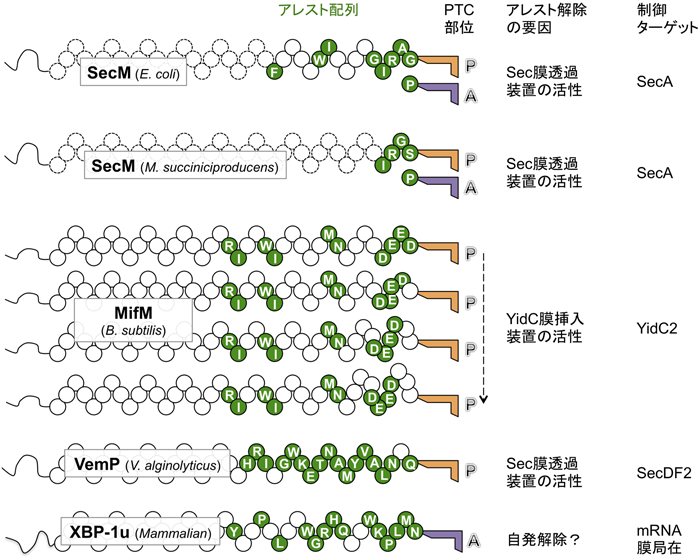
翻訳アレストを起こすアミノ酸配列はSecM以外にも,種々の生物で見つかっている13).アレストに重要なアミノ酸残基と停止したリボソームの状態が解明されている代表的な例を図3,図4に示した.各論は別の総説13)に譲り,ここではアレストペプチド研究からどのような意味合いを汲み取ることができるかを考察する.翻訳アレストを起こすアミノ酸配列は,3残基から10残基以上まで長さに幅があり,配列自体に共通性がない.アレストした翻訳複合体の構造解析31,42–46)によれば,翻訳途上鎖とリボソームの構成成分の近接・相互作用の仕方も個別性が高い.リボソームの変異が翻訳アレストに与える影響も,それぞれのアレストペプチドごとに異なっている.多様なアレスト配列は,進化の上でも比較的最近につけ加わったようだ.たとえば,SecMはγproteobacteria網のEnterobacteriales目とPasteurellales目だけに47),膜組込み装置のモニター基質であるMifM(後述)はFirmicutes網のBacillales目だけに48)存在する.リボソームトンネルと相互作用できる配列が多数ありうる中で,生物はたまたま一つのアレスト配列を利用しているように思われる.大腸菌ではSecAがSecMで制御されるのに対して,海洋ビブリオ属ではSecDFホモログの発現がuORFであるVemPにおける翻訳アレストによって制御されることが最近明らかになった49).進化的に保存された基幹的な装置の制御が,いわば後づけの制御機構ともいえるアレストペプチドによってなされることは,生物の多様性獲得において意味があることかもしれない.アレストペプチドが「後づけで」容易に出現しうることは,人工的なアレスト配列を作り出すことが容易にできることからも示唆される12).種特異的なアレスト配列も存在する.MifMの翻訳伸長アレストは枯草菌のリボソームで強く起こるが,大腸菌では微弱にしか起こらない50).この種特異性は,L22の1残基の変異で解消するような微妙なものである42).進化的保存性が高いリボソームが種特異的なタンパク質をも首尾よく合成(あるいはアレスト)するにあたり,リボソームトンネルによる新生鎖識別能が微調整されてきたのであろう.プロテオームとリボソームトンネルは微妙な相互関係の下に進化してきたことが想像される.
一方で,合成途上ポリペプチドのアミノ酸配列の点検に共通性の高いリボソーム成分が関与することも事実である(図2).すでにSecMの説明で登場した,トンネル狭窄部位のL22(真核細胞ではL17),rRNA残基A751(大腸菌リボソームの残基番号で表示),L4,rRNA残基A2058など13),PTC付近のrRNA残基A2062やU2585などである.アレストペプチドの残基との相互作用の結果PTC塩基が異常な配向をとり,P部位やA部位のtRNAの配向異常や格納不全が生ずる31,42–46).
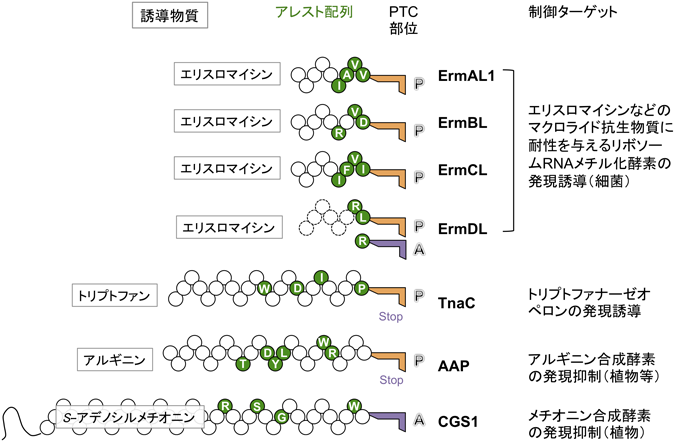
アレスト状態のペプチジルtRNAは,P部位に存在する場合が多い(図3,図4).A部位のアミノアシルtRNAに特異的なアレストとA部位のアミノアシルtRNAには依存しないアレストが知られる.P部位のアミノ酸の種類にすら依存しないアレストもある.この最後のカテゴリーでは,トンネル領域での合成途上鎖—リボソーム相互作用によって,何らかのアロステリックな構造変化が起こりPTCが不活化するのだろう.このタイプに属するMifMは,連続した4か所でアレストする(図3).このユニークなマルチサイトアレストには,アレスト部位の直前に酸性アミノ酸クラスターが存在することが重要である51).逆に,合成途上鎖がトンネル内部に届かない段階でのアレストも知られる.ErmDLは,3アミノ酸Arg-Leu-Argのみで抗生物質に依存した(後述)伸長阻害をLeuとArgの間で起こす.そのため,最短ではMet-Arg-Leu-tRNAがP部位に,Arg-tRNAがA部位に存在するアレスト複合体を作ることができる(図4)52).翻訳終結ステップでアレストを起こす例としてTnaCやAAPがあり,アレスト誘導分子であるアミノ酸存在下で翻訳終結が阻害される(図4)43).翻訳における転座(translocation)ステップでアレストを起こす例としてCGS1があり,合成途上ペプチジルtRNAがA部位に存在する状態でアレストする(図4)53).このように,さまざまな合成途上ポリペプチドが翻訳のさまざまな過程を損なう.
2)翻訳アレストの制御
mRNA上にリボソームが常に立ち止まっていることは細胞にとって不都合である.通常,アレストペプチドによる翻訳アレストは必要なときにしか起こらないように制御される.方向性が異なる二つの制御様式がある13).SecM,MifM,VemPのアレスト配列のように,単独で翻訳阻害能力を持つ内在性(intrinsic)アレスト配列はアレスト解除機構によって制御され(図3),特定の低分子物質が存在するときに限って翻訳アレストを起こす誘導性(inducible)アレスト配列は,誘導物質の濃度によって制御される(図4).
ⅰ)内在性翻訳アレストの制御
すでに論じたようにSecMの合成途上鎖は膜透過によりアレスト解除される.海洋性ビブリオのVemPにおける伸長アレストもSecMと同様,Sec膜透過装置によって解除される(図3)49).MifMはN末端にYidC膜挿入装置54)の基質となる疎水性膜挿入配列を持ち,合成途上鎖の膜挿入に共役してアレストが解除される(図3)48).この場合にも,新生鎖に加わる物理力がきっかけとなることが考えられる.MifMのN末端膜挿入配列を分泌タンパク質のシグナル配列と置き換えると,Sec膜透過によってアレストが解除されるようになる50).アレスト解除への物理力の関与と合致すると同時に,アレスト配列とモニター部分の組み合わせによって,調節性新生鎖が出現したとの考えにつながる.ただし,SecMのアレスト解除メディエーターのような第三の要素による仲立ちによって,N末端のモニター部位が効率よくアレスト解除を誘起できる可能性は残る.
ウイルスの遺伝情報発現に特化した仕組みであるUL4ORF255)における翻訳アレストや2Aペプチド56)翻訳で起こるかもしれないアレスト(後述)は解除機構が存在しない一過性のものかもしれない.同様に自発的に解除される例として,XBP-1uでみられる翻訳アレスト57)(後述)がある.
ⅱ)誘導性翻訳アレストの制御
誘導性の翻訳アレストにおいて,uORFによってコードされるアレストペプチドが特定の低分子の存在下で翻訳アレストを起こし,標的遺伝子の発現を正(細菌の場合)または負(真核細胞の場合)に制御する(図4)13).誘導物質がリボソームトンネルの内部に入り込んで働くとすれば,リボソーム外に制御ドメインを持つ必要はなく,誘導性アレストペプチドは短いものが多い(図4).細菌の抗生物質耐性遺伝子が抗生物質によって発現誘導される現象はアレストペプチドによる発現制御機構として最初に発見された.クロラムフェニコールによるアセチル基転位酵素(グラム陽性菌)や多剤排出ポンプ(グラム陰性菌)の翻訳誘導58),マクロライド系抗生物質によるリボソームRNAメチラーゼ(耐性をもたらす)の翻訳誘導が知られる.後者では,リーダーペプチドErmAL,ErmBL,ErmCL,ErmDLなどが翻訳アレストを起こす59).A,B,C,Dそれぞれの系は,マクロライド系抗生物質による誘導の特異性が異なり,アレストに必要なアミノ酸配列,リボソームにおける構造とアレストの分子機構もそれぞれ異なっている44,45,52,60–62).同様の機能を持つErmリーダーペプチドの個別性から,アレストペプチドの多様性と後発進化が再確認される(図4).マクロライドはリボソームトンネルの入り口付近のrRNA残基A2058—A2059付近に結合する.ErmCLのアレスト配列自体は4残基であるが,合成途上鎖の長さ9残基がアレストの誘導には必要である.よって,薬剤はアレストペプチドとリボソームの両方と相互作用して翻訳アレストを誘導するのだろう.一方,ErmDLの系では,薬剤がリボソームのアロステリックな構造変化を誘起して,ErmDL自体と直接相互作用することなく,アレストを誘導する.従来,マクロライド系抗生物質は,リボソームトンネルを塞いで無差別的に翻訳伸長を阻害するといわれていたが,実際には,リボソームの合成途上鎖識別機能に働きかけて配列特異的な伸長阻害を引き起こし,タンパク質合成のバランスを崩すことにより抗菌作用を示すと考えられるようになった63).Ermリーダーペプチドはこのようなリボソームと抗生物質の性質をうまく利用している.
トリプトファン64),アルギニン65),ポリアミン66),S-アデノシルメチオニン53)などもアレスト誘導物質として知られる(ポリアミンがアレスト解除の方向に働く系もある67)).ショ糖68),アスコルビン酸69)が働く系もありそうだ.大腸菌のトリプトファナーゼ–トリプトファン透過酵素オペロンのuORFの産物TnaCは,トリプトファン存在下で翻訳終結アレストを起こし,オペロンの転写を促進する(図4)64).最近の構造決定によるとTnaC新生鎖がrRNAと相互作用して疎水性の溝を2か所形成しトリプトファン2分子を収容する43).このトリプトファン結合部位はマクロライド抗生物質の結合部位とオーバーラップした,残基U2609,A752,A2058,A2059などが取り囲むリボソーム内のスペースにある.リボソームのこの領域は低分子物質を感知する領域かもしれない.TnaC合成途上鎖はL22の残基とも相互作用し,これら相互作用の総和としてPTC反応に重要なU2585やA2602残基の配向を翻訳終結因子RF2が働けない配向に固定してしまうのであろう.
3)翻訳アレストのアウトプット
アレストペプチドの翻訳アレストがもたらす生物学的効果は,「翻訳アレストに伴いmRNAの状態変化が起こり,生物学的な効果が生ずる」と一般化できる13).その要因となるのがmRNA上で立ち止まったリボソーム,あるいは合成途上鎖のリボソームの外部に出た部分の挙動である.
ⅰ)標的遺伝子の発現誘導
細菌で典型的な効果は,リボソームの停滞によるmRNA二次構造の変化(SD配列の露出)による下流の標的遺伝子の翻訳誘導であり,secM-secA制御系について詳しく述べた.ビブリオ菌が低塩環境にさらされるとNa+イオン駆動性のSecDF1が不活化されて膜透過活性が低下する.VemPはそれを感知した翻訳アレストによって,プロトン駆動性のSecDF2パラログの翻訳を誘導する49).枯草菌では,YidC膜挿入装置の活性が低下すると,YidCホモログの一つYidC2のuORF産物MifMにおける翻訳伸長アレストによって,YidC2の翻訳が誘導される48).Ermリーダーペプチドにおける翻訳アレストも同様の機構によって,抗生物質耐性遺伝子の翻訳を誘導する59).TnaCの場合には,リボソーム停滞によるmRNA二次構造の変化によって,転写終結因子ρが働けない状態となり,トリプトファナーゼとトリプトファン透過酵素の転写が促進される70).
ⅱ)標的遺伝子の発現抑制
真核細胞ではuORFでのリボソームの停滞は,負の制御をもたらす(図4).立ち止まったリボソームが標的遺伝子の翻訳開始コドンへのリボソームの流れ(スキャニング)を断ち切ることにより,標的遺伝子の翻訳を抑える13,71).たとえば,AAPはアルギニン依存の翻訳終結アレストを起こすことにより,アルギニン生合成酵素の翻訳を抑制する65).また,ポリアミン合成に働くS-アデノシルメチオニンデカルボキシラーゼの発現はポリアミン誘導性のuORFの翻訳終結アレストにより抑制される66).ヒトサイトメガロウイルスの糖タンパク質UL4遺伝子の発現は,uORFにおける翻訳終結アレストによって抑制される55).植物のメチオニン生合成に働くCGS1(cystathione-γ-synthase)は,uORFではなく酵素自身の一次構造の途中にアレスト配列を含む(図4)53).S-アデノシルメチオニン誘導性の転座アレスト(図4)によって,酵素合成が直接中断される.加えて,立ち止まったリボソームの5′側でのmRNAの切断が誘導されて,強固なフィードバック抑制が達成される.
ⅲ)翻訳アクロバット・リコーディングの誘導
翻訳の停滞により,翻訳系の特殊な働きが誘導されることがある.プログラムされたフレームシフトや翻訳のホッピングは翻訳伸長停滞により促進され72),停滞には合成途上鎖のアミノ酸配列が関与する場合もある73).手足口病ウイルスの2Aペプチドとその下流のペプチドは特殊な翻訳終結と再開始によって生じる56).このStopGo翻訳が起こるにあたって,翻訳が一時的に停滞するとの報告がある74).
ⅳ)翻訳装置,翻訳産物の品質管理
D-アミノ酸が合成途上ポリペプチドに取り込まれると,合成途上鎖がPTCを阻害して翻訳伸長不全に至るとの報告がある75).異常なアミノ酸を含むタンパク質合成を防ぐ品質管理機構として翻訳アレストが使われる例といえるかもしれない.真核細胞で知られる,連続する塩基性アミノ酸での翻訳アレスト76)やポリアミン誘導性のuORFの翻訳アレストも品質管理機構の対象となる77).真核細胞におけるuORFは対象遺伝子を負に制御するため,品質管理機構によるmRNAの分解は目的にかなっている.細菌のアレストペプチドによる正の発現制御が直ちにはリボソームレスキュー系による処理を受けないこと20,21,78)と対照的である.
ⅴ)mRNAの局在化
XBP-1u mRNAは,小胞体ストレス応答に際して,小胞体膜に局在する酵素によるスプライシングを受けて転写因子をコードできるようになる.効率よいスプライシングのため,このmRNAは,自らがコードする翻訳途上鎖の疎水性領域が膜に結合することを利用して膜に局在化する79,80).この疎水性領域がリボソームの外に出現した状態で翻訳がアレストするため(図4),mRNAが膜近くに局在できる57).アレストは自発的に解除され,スプライス型mRNAが完成する.合成途上鎖を介するmRNA局在化機構は一般化できそうだ.我々は,合成途上SecMにはsecM-secA mRNAを膜に局在化する役割があると考えている.膜環境での合成により新生SecAの活性構造へのフォールディングが促進される可能性を示唆する実験結果があるからである81).翻訳途上鎖は,タンパク質合成の「場所取り役」も務める.
4)翻訳と構造形成との微妙な相互関係
アレスト配列の1アミノ酸置換変異の中には,完全なアレスト欠損を示すもの以外に,種々の程度に部分欠損を生ずるものがある22).すなわち,アミノ酸配列は,微弱なポーズによって翻訳伸長速度を微調整することができる.このような翻訳のブレーキが,翻訳途上鎖のターゲティング,フォールディング,修飾などに寄与することが考えられる.すでに論じたように,リボソームの外(細胞質)に出現した直後,あるいはトンネル内部でのドメインフォールディングが翻訳アレスト解除の方向に働く(図5)28,38).翻訳途上鎖とリボソームがフィードバックループを形成して,緩急のリズムをつけながらタンパク質ができあがっていくという考えが現実味を帯びてきた.Puglisiは,「翻訳とフォールディングの微妙なダンス」という表現でこのような概念を解説した82).
5)翻訳の応答性と自律性
最近,熱ストレス条件下やHsp70シャペロン系の機能不全時に,種々のタンパク質の翻訳伸長がN末端に近い領域で停滞するという報告がなされた83–85).正常時には,合成途上鎖のフォールディングやHsp70の結合が翻訳伸長を助けている可能性が浮上したのである.本稿で議論してきたように,翻訳は機械的で単調な各ステップの繰り返しで進行するものではなく,リボソームは常に自分が合成する産物を吟味して,局所的にスピードを微調整している.一方で,産物である翻訳途上鎖はリボソームから生まれ落ちる前に,細胞社会の影響を受け始める.そして翻訳途上鎖のダイナミックな動きや代謝産物の変動が翻訳のステップそのものに制御をかける.翻訳は環境にも応答できるダイナミックなプロセスであるという特筆すべき事実が垣間みえてきたように思われる.これらは天文学的なスケールの配列情報を担う合成途上鎖とそれらを創って送り出すリボソームの二つが主役になって演じられる自律的な4次元ドラマである.我々は現在,セントラルドグマが分子の自律性によって制御されるという新たなパラダイムを獲得しつつあるのではないだろうか?