獲得免疫を担うT細胞およびB細胞が発現する抗原受容体は,VDJ組換えと呼ばれる遺伝子の再構成によってほぼ無限の多様性を獲得する一方,その構造に異常があり無用なものや,運悪く自己抗原に対して高い親和性を持つ有害なものが生み出される可能性がある.T細胞の場合は胸腺において,構造に異常があるT細胞抗原受容体(T cell receptor: TCR)を発現するT細胞は十分なTCR刺激を受けられずに,また自己反応性TCRを発現するT細胞は過剰なTCR刺激を受けて,いずれもアポトーシスにより除去される.有用なT細胞を選別する前者が正の選択(positive selection),有害なT細胞を排除する後者が負の選択(negative selection)と呼ばれる.PD-1(programmed cell death-1)は1992年に,京都大学の本庶および石田(現・奈良先端大)らによって,自己反応性T細胞にアポトーシスを誘導する分子の候補として同定されたⅠ型膜タンパク質である1).その後の解析から,PD-1は胸腺ではなく末梢において,また除去するのではなく無力化することによってではあるものの,自己反応性T細胞の抑制に関与することが明らかとなった2–4).また,自己免疫に加えて,感染免疫,腫瘍免疫,移植免疫等,さまざまな免疫応答を抑制することが明らかとされている.
がんの治療において,免疫療法は外科療法,化学療法,放射線療法に続く第4の治療法として期待され,がん細胞に対して免疫応答を誘導あるいは増強する試みが精力的に行われてきたが,期待されるほどの効果は得られていなかった.むしろ炎症反応によりがん化が促進されるという実験結果が得られる等,長らくがんと免疫の関係は混沌としていた.近年,がん細胞が無秩序な増殖能を獲得して増大する過程において,免疫系の監視を受け,免疫監視から逃避する術を獲得したものが選択されているとする,免疫編集(immuno-editing)という概念が提唱されている5, 6).PD-1による腫瘍免疫の抑制,PD-1阻害抗体による腫瘍免疫の増強,さらには免疫応答の誘導に頼った従来型免疫療法の困難さは,まさしく免疫編集の概念を支持しているといえる.
本稿では,これまでに明らかとなっている自己免疫およびがん免疫におけるPD-1の機能について概説する.
PD−1は,培養T細胞株2B4.11が強いTCR刺激によりアポトーシスする際に発現が増強する遺伝子として,サブトラクティブハイブリダイゼーション法にて同定された,Ⅰ型膜タンパク質をコードする遺伝子である1).未刺激の2B4.11細胞株にPD-1遺伝子を強制発現させてもアポトーシスが誘導されなかったことから,その機能は長らく不明であったが,後述のとおり,PD-1欠損マウスが自己免疫疾患を自然発症したことから免疫応答の制御因子であることが示唆されるとともに,分子機序の解析により,PD-1が抗原受容体刺激を負に制御する抑制性の補助受容体であることが明らかとなっている7–9).
PD-1は,T細胞の活性化における興奮性の補助受容体CD28および抑制性の補助受容体cytotoxic T lymphocyte antigen-4(CTLA-4)と20~30%のアミノ酸相同性を有しており,ファミリーを形成する.PD-1は,未刺激の細胞には発現しておらず,T細胞,B細胞,NK細胞,および骨髄系細胞において,活性化に伴って発現が誘導される.また,抗体産生においてB細胞の分化を促進する濾胞ヘルパーT細胞上には恒常的に発現している10).さらに,慢性ウイルス感染症において慢性的な抗原刺激により機能が減弱した疲弊T細胞,老化に伴い出現する機能が制限された老化T細胞等においても,恒常的にPD-1が発現している11–13).一方,CD28とCTLA-4の発現はT細胞に限局される.CD28が抗原に遭遇したことがない未感作のナイーブT細胞にも発現しているのに対して,CTLA-4はナイーブT細胞には発現しておらず,活性化により発現が誘導される.
CD28およびCTLA-4との類似性から,PD-1のリガンドは,CD28およびCTLA-4のリガンドであるB7.1(CD80)およびB7.2(CD86)と相同性を有すると予測された.実際,PD-1との結合能を指標にしたB7様分子の探索により2種類のリガンドが同定され,PD-L1およびPD-L2と命名された14,15).
PD-L1は,樹状細胞,リンパ球,マクロファージ,肥満細胞等,抗原提示細胞を中心とした多くの免疫系細胞に発現が認められ,サイトカイン等の刺激により発現量が増加する.PD-L2の発現は樹状細胞,マクロファージ,肥満細胞等に認められ,やはりサイトカイン等の刺激により発現量が増加する16,17).PD-L1の発現は,免疫系細胞に加え,心臓,肺,肝臓,胎盤,角膜等の末梢実質臓器にも認められ,やはりサイトカイン等の刺激により発現量が増加する.胎盤や角膜は,免疫応答が制限されておりimmune-privilege(免疫特権)を有すると考えられているが,免疫系からの隔絶にPD-L1が関与している可能性が示唆されている18,19).一方,末梢実質臓器におけるPD-L2の発現は限定的である.また,一部のがん細胞株やヒトがん組織,ウイルス感染細胞において,PD-L1およびPD-L2の発現が認められる.B7の発現が主に抗原提示細胞に限局していることから,PD-1とCTLA-4は空間的に使い分けられていると考えられる.
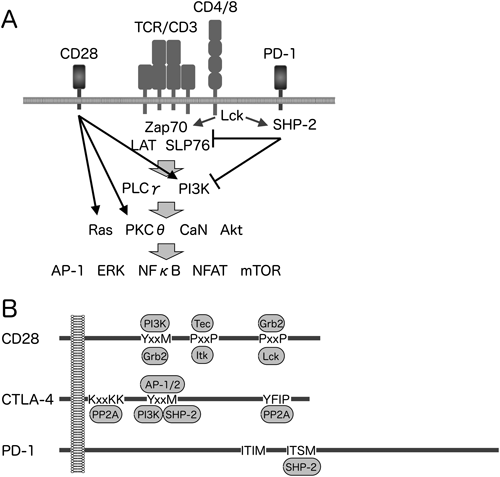
主要組織適合抗原複合体(MHC)に提示される抗原をTCRが認識すると,Srcファミリーに属するチロシンキナーゼLckが活性化される.TCRと複合体を形成するCD3分子の細胞内領域に存在するITAM(immunoreceptor tyrosine based activation motif)のチロシン残基をLckがリン酸化し,そこに別のキナーゼであるZAP70がSH2ドメインを介して結合する.ZAP70はリンカーであるSLP76やLATをリン酸化し,さらに下流のシグナル伝達分子がリクルートされる.これにより,NFκB,NFAT,MAPK等が活性化され,細胞増殖や機能分化に関与する分子,各種サイトカイン等の発現が誘導される(図1)4,20).
CD28およびCTLA-4は,免疫グロブリン可変部(IgV)様ドメイン,膜貫通領域および細胞内領域からなる.T細胞の抗原刺激時にCD28がB7.1あるいはB7.2と結合すると,CD28の細胞内領域にPKCθ,PI3K等がリクルートされ,抗原受容体刺激が増強される.一方,CTLA-4が同時に発現する場合には,B7.1およびB7.2に対する親和性がCD28よりもCTLA-4の方が約10倍高いため,CTLA-4がB7をCD28から奪い,CD28からの興奮性補助シグナルが阻害される.またCD28の競合阻害に加え,CTLA-4の細胞内領域にタンパク質脱リン酸化酵素であるSH2-containing protein tyrosine phosphatase-2(SHP-2)等がリクルートされ,抗原受容体刺激を積極的に抑制するシグナルが伝達される4,20).
PD-1は細胞内領域にチロシン残基を含むシグナル伝達モチーフであるITIM(immunoreceptor tyrosine-based inhibitory motif)とITSM(immunoreceptor tyrosine-based switch motif)を有している.TCR刺激時にPD-1がPD-L1あるいはPD-L2と結合すると,SrcファミリーキナーゼによりPD-1のITSMがチロシンリン酸化を受け,このリン酸化チロシンにSHP-2がSH2ドメインを介して結合する.興味深いことに,ITIMのチロシン残基もリン酸化を受けるがSHP-2とは会合しない.抗原受容体刺激により活性化されたキナーゼによってリン酸化を受けた,ZAP70をはじめとした各種シグナル伝達分子のチロシン残基を,SHP-2が脱リン酸化することにより,抗原受容体刺激が抑制されると考えられている9,21,22).
ITSMのswitchという名称は,CD150のITSMがSH2D1A(SAP)存在下では活性化のシグナルを伝達し,非存在下では抑制性のシグナルを伝達することに由来するが,これまでのところ,PD-1はSAPと結合しないと報告されており,PD-1のITSMがswitchして興奮性のシグナルを伝える可能性については否定的である.
T細胞が抗原提示細胞によって活性化される際,接着面に神経細胞間で形成されるシナプスと似た構造が形成され,免疫シナプスと呼ばれる.免疫シナプスは,TCR複合体,接着分子,膜型受容体等が同心円状に配置された構造であり,TCR刺激の足場として機能する.横須賀らは,全反射蛍光顕微鏡を用いて,PD-1の免疫シナプスにおける機能を明らかにしている22).T細胞が抗原提示細胞と接触すると,数十秒のうちに30~150個のTCR複合体からなるマイクロクラスターが100~200個形成される.その後,数分間のうちにTCR複合体マイクロクラスターが接着面の中心部に移行することにより,免疫シナプスが形成される.PD-1は,TCR複合体マイクロクラスターに入り込み,活性化シグナルを抑制して免疫シナプスの形成を阻害することにより,T細胞と抗原提示細胞の接着を不安定化させる.一方,CD28はTCR複合体とマイクロクラスターを形成するが,免疫シナプス形成時には,TCR複合体の外側を取り囲む位置へと移動する.CTLA-4は,細胞質内の小胞内に存在し,刺激により細胞表面上に表出してくるため,抑制のタイミングがPD-1よりも遅く,TCR複合体マイクロクラスターが凝集した後に,本来CD28が位置するTCR複合体の外輪部を占拠してCD28シグナルを遮断する23).今後,各種興奮性および抑制性補助受容体がどのように協調して,あるいは独立に各種免疫応答を制御しているのかが明らかとされるとともに,作用機序に基づいた最適な制御方法が開発されることが期待される.
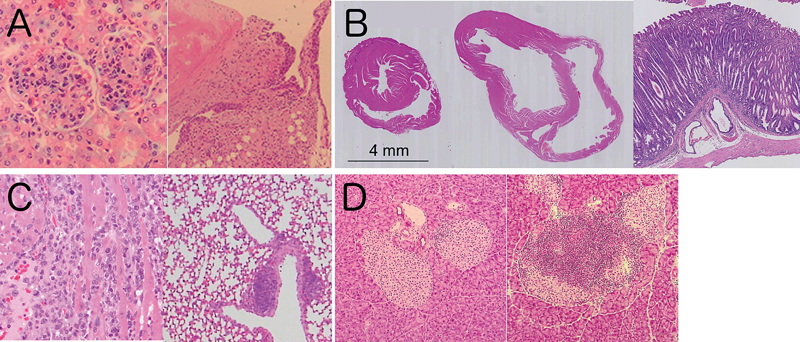
PD-1欠損マウスは,129/Sv系統由来のES細胞を用いて作製された後,C57BL/6系統に戻し交配されたが,当初は明確な自己免疫疾患の発症は確認されなかった7).その後,C57BL/6系統への戻し交配を繰り返して遺伝子背景を均一化するとともに,加齢時の影響を詳細に観察することにより,SLE様の糸球体腎炎および関節炎の発症が確認された(図2A)8).興味深いことに,PD-1欠損マウスを他の系統に戻し交配して遺伝子背景を変更すると,発症する自己免疫疾患の標的臓器と病態が変化した.BALB/c系統に戻し交配したPD-1欠損マウス(BALB/c-PD-1欠損マウス)は自己免疫性の拡張型心筋症および胃炎を自然発症した(図2B)24,25).MRL-PD-1欠損マウスは激しい心筋炎,肝炎,肺炎,胃炎等を自然発症し,約80%のマウスが生後10週齢までに死亡した(図2C)26).また,Ⅰ型糖尿病のモデルマウスであるNODマウスにPD-1欠損を導入すると,Ⅰ型糖尿病の発症が大幅に促進された(図2D)27).また,NODマウスは唾液腺炎を自然発症することから,シェーグレン症候群のモデルマウスとしても使用されているが,PD-1欠損により唾液腺炎も増悪されるものの,他の臓器における非特異的な炎症は認められず,自己免疫応答の臓器特異性は保たれていた.
ヒトにおいても,PD-1遺伝子の多型が全身性エリテマトーデス,関節リウマチ,Ⅰ型糖尿病等,複数の自己免疫疾患の発症や病態に関連することが報告されているが,人種によって,関連を示す疾患,関連の強度等が大きく異なる2).このように,遺伝背景により病態が変化することは,PD-1が自己免疫寛容において普遍的に重要な制御分子であることを示唆するとともに,PD-1が他の分子と協調して自己免疫疾患の発症を制御していることを示唆する.協調して働く分子の機能あるいは組合わせが遺伝背景や環境によって異なるため,異なる遺伝背景や環境によって異なる種類の自己免疫疾患を発症するものと考えられる.
標的臓器に加えて,病態の発症メカニズムもマウスの遺伝子背景により大きく異なる.BALB/c-PD-1欠損マウスにおける拡張型心筋症では,心筋型トロポニンⅠに対する自己抗体が産生され,この抗体が心筋細胞のカルシウム電流を増加させることにより心筋細胞を疲弊させ,心筋が収縮不全を来す28).また,胃炎の発症も,胃壁細胞に特異的な自己抗体の産生を伴う.一方,NODマウスにおけるⅠ型糖尿病の発症においては,ランゲルハンス島へのTリンパ球の浸潤に伴って,β細胞上にPD-L1の発現が誘導されるが,β細胞上のPD-L1が自己反応性Tリンパ球を直接抑制することによりⅠ型糖尿病の発症を遅延させる27).MRL-PD-1欠損マウスにおける心筋炎,後述のNOD.H2b-PD-1欠損マウスにおける末梢神経炎においても,標的細胞におけるPD-L1の発現誘導が認められることから,PD-L1の発現誘導は,自己反応性T細胞による傷害からの回避を目的として,複数の臓器で用いられている機構であると考えられる26).
PD-1は標的細胞の傷害や自己抗体の産生を抑制するのに加え,自己反応性T細胞にアナジー(免疫不応答)を誘導することにより,自己免疫疾患の発症を抑制する29).また,PD-1シグナルにより誘導性の制御性T細胞が産生されるという報告もあるが30),各疾患の発症制御においてPD-1のこれらの機能がどのように関与しているのかについては,今後より詳細な検討が必要であると思われる.いずれにせよ,用いる実験系や患者ごとにPD-1の作用機序が異なる可能性があるため,各種疾患の治療を目的としてPD-1およびPD-1関連分子の機能を人為的に操作する際には,その影響を多角的に検証し,正確に理解することがきわめて重要である.
6. PD-1欠損マウスの症状に遺伝素因が与える影響
PD-1はさまざまな環境において,さまざまな分子と協調して自己免疫疾患の発症を制御していると考えられるが,PD-1と協調して働く分子を特定することができれば,自己免疫疾患の発症を制御する多因子ネットワークシステムの全貌が解明できるとともに,自己免疫疾患やがんに対して,より効果的な治療法を開発できると期待される.
NODマウスを用いたⅠ型糖尿病の連鎖解析は,これまでに多くのグループによって精力的に行われており,20以上のIdd(insulin-dependent diabetes)遺伝子座が同定されている31).しかし,あまりにも多くの遺伝子座がⅠ型糖尿病の発症に関与するため,各遺伝子座がどのように関与して実際にⅠ型糖尿病が発症するかを解析することはきわめて困難であり,ほとんど進んでいない.NODマウスのⅠ型糖尿病発症頻度には性差があり,また飼育環境によって発症頻度が変化する.より発症頻度が高いメスでは,15週齢ごろよりⅠ型糖尿病を発症するマウスが現れ,40週齢までに40~70%のマウスがⅠ型糖尿病を発症する.PD-1欠損下では,オスメスともに4週齢ごろよりⅠ型糖尿病を発症するマウスが現れ,10週齢までにすべてのマウスがⅠ型糖尿病を発症したことから,PD-1欠損マウスを用いることにより,より感度が高い連鎖解析を行うことができると期待された.NOD-PD-1欠損マウスとC57BL/6-PD-1欠損マウスの交雑F2およびBC1マウスを用いた解析では,4染色体領域のみが連鎖を示した27).免疫応答の増強に関わる多くのIdd遺伝子座の影響がPD-1欠損によって代償されたためと考えられる.また,これら4染色体領域については,PD-1欠損によって代償されないことから,Ⅰ型糖尿病により特異性が高い影響を有すると考えられる.
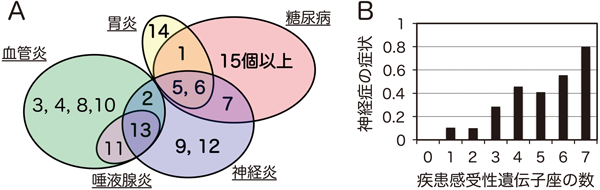
これら4染色体領域のうち,MHC遺伝子を含むIdd1領域が最も強くⅠ型糖尿病の発症に連鎖を示すが,NODマウスのMHCは,ヒトにおいてⅠ型糖尿病の発症と連鎖を示すMHCとよく似た特徴を有しており,g7ハプロタイプと呼ばれている.Ⅰ型糖尿病に抵抗性があると報告されているbハプロタイプに置換したNOD.H2b-PD-1欠損マウスは,Ⅰ型糖尿病をまったく発症せず,ヒトのギラン・バレー症候群や慢性炎症性脱髄性多発神経炎に類似した末梢神経炎を自然発症した(図3)32).
NOD.H2b-PD-1欠損マウスとC57BL/6-PD-1欠損マウスの交雑F1マウスをNOD.H2b-PD-1欠損マウスに戻し交配して得られたBC1マウスでは,約20%のマウスに末梢神経炎,約49%のマウスに唾液腺炎,約66%のマウスに胃炎,約40%のマウスに腎血管炎,および約14%のマウスにランゲルハンス島炎が認められた(図3).連鎖解析により,末梢神経炎,唾液腺炎,胃炎,腎血管炎およびランゲルハンス島炎の発症に連鎖を示す遺伝子座が,各々7個,2個,4個,7個および1個,同定された33).図4に示すとおり,胃炎とランゲルハンス島炎の感受性遺伝子座はⅠ型糖尿病感受性遺伝子座として報告されている遺伝子座とほぼ一致したが,血管炎と唾液腺炎の感受性遺伝子座は,Ⅰ型糖尿病感受性遺伝子座とまったく一致しなかった.一方,末梢神経炎感受性遺伝子座は,Ⅰ型糖尿病感受性遺伝子座と血管炎・唾液腺炎感受性遺伝子座と重複する領域,およびそれらとは重複しない領域に存在していた.すなわち,NODとC57BL/6というわずか2系統のマウスの染色体だけでも,さまざまな疾患に対して感受性あるいは抵抗性を有する多型を数多く有しているといえる.また,多遺伝子疾患の特徴として,これらの感受性遺伝子座の数と,疾患発症の頻度や症状が正の相関を示すとともに,ほとんどの遺伝子座が抵抗性の多型であっても,感受性遺伝子座を1個有するだけで,低いながらも一定の頻度で疾患を発症することが確認された(図4B).このことは,後述のPD-1阻害抗体による副作用を考える上で,きわめて重要な意味を持つといえる.
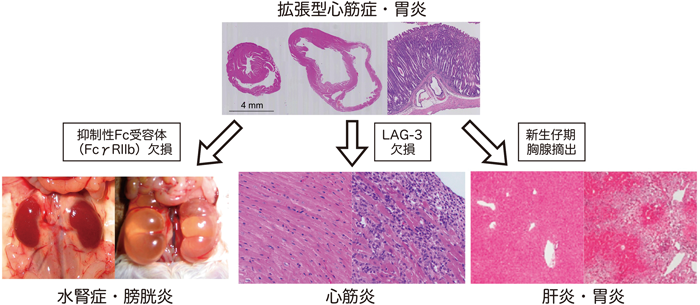
B細胞抗原受容体遺伝子は,発生段階のVDJ組換えとは別に,抗原刺激により体細胞突然変異とクラススイッチ組換えという2種類の遺伝子変換を受け,抗原に対する親和性が増強するとともに,抗体のクラスがIgMからIgG等へと変化して機能が多様化する34,35).BALB/c-PD-1欠損マウスが発症する拡張型心筋症と胃炎においてはIgGクラスの組織特異的な自己抗体が検出されるが,体細胞突然変異とクラススイッチ組換えに必須の酵素であるAID(activation-induced cytidine deaminase)を欠損させると,拡張型心筋症と胃炎の発症が認められない36).一方,抑制性のFc受容体であるFcγRIIBを同時に欠損させても,拡張型心筋症と胃炎の発症頻度は変化しなかったが,約36%のBALB/c-PD-1・FcγRIIB二重欠損マウスが腎盂,尿管,および膀胱の移行上皮に特異的な自己抗体の産生を伴う両側性の水腎症および間質性膀胱炎を自然発症した(図5)25).このことは,FcγRIIBによる抑制の程度が,標的臓器により異なることを示唆する.
ヘルパーT細胞の補助受容体CD4の近縁分子として1990年に同定されたLAG-3は,CD4を競合阻害することにより,T細胞の活性化を抑制すると報告されている.BALB/c-PD-1欠損マウスにおいて,LAG-3を同時に欠損させると,激しい心筋炎を発症して生後10週齢までに死亡した(図5)36).発現時期,抑制機能等から,LAG-3が最初に自己反応性Tリンパ球を抑制し,それでも抑制できなかった場合にはPD-1が二重安全装置として働くものと考えられる.また,Wooらによって,LAG-3とPD-1が腫瘍免疫においても協調的に働くことが報告されている37).
また,BALB/c-PD-1欠損マウスにおいて,生後3日目に胸腺を摘出して制御性T細胞不全を誘導すると,自己免疫性の肝炎を発症して早期に死亡した(図5)38).これらのことから,PD-1はさまざまな免疫制御分子と,各種免疫応答を協調的に,あるいは独立に制御することにより,自己免疫疾患の発症を抑制していると考えられる.今後,協調作用の分子メカニズムおよび他の免疫現象における各種分子の協調作用をより詳細に解析することにより,自己免疫寛容の成立・維持に関わるネットワークシステムの全貌が明らかになると期待される.
がん細胞は正常細胞から発生するため,がん免疫は一種の自己免疫と考えることができる.また,前述のとおりPD-1欠損マウスに発症する自己免疫疾患の解析から,標的自己細胞がPD-L1を発現することにより自己反応性T細胞の傷害を回避していると考えられたが,培養がん細胞株の中にPD-L1あるいはPD-L2を恒常的に発現しているものが多数認められたことから,PD-1によってがん免疫応答も抑制されている可能性が考えられた.肥満細胞腫株P815をBALB/cマウスの皮下に移植すると腫瘍が形成されるが,PD-L1を強制発現させたP815細胞株(P815-PD-L1)では,形成される腫瘍が増大していた.また,PD-1シグナルを抗PD-L1抗体を用いて阻害することにより,腫瘍の形成や増大が阻害されることが確認された.さらに,P815-PD-L1細胞株はP815細胞株と比較して,細胞傷害T細胞による細胞傷害に抵抗性であった.PD-1シグナルの阻害による腫瘍の縮小は,PD-L1を恒常的に発現するミエローマ細胞株J558Lを用いた実験でも確認されたが,PD-L1をほとんど発現しないメラノーマ細胞株B16では認められなかった39).これらの結果から,がん細胞上のPD-L1がT細胞上のPD-1を介して抑制シグナルを入れ,細胞傷害活性を減弱することにより,生体の抗腫瘍免疫応答から逃避していることが明らかとなった.また,定常状態ではPD-L1を発現していないB16細胞株についても,脾臓に移植して肝臓への転移を解析する実験系では,生体内でPD-L1の発現が誘導されてPD-1シグナルの阻害に感受性となり,肝臓への転移が強力に抑制された40).したがって,PD-1阻害による抗腫瘍免疫応答の増強効果は,がん細胞の特性だけでなく,発生母地や転移先臓器の特性に大きく影響を受けると考えられる.また,PD-L1の発現が環境によって変化することから,ある状態におけるPD-L1の発現だけでは,PD-1阻害抗体の治療効果を予測することは困難であることが示唆された.
PD-L1が報告される数か月前に,Chenらのグループによって,同じ分子が受容体未知の新規B7ファミリー分子としてB7-H1という名前で報告されているが,彼らは,未知の受容体を介して興奮性の免疫補助シグナルを伝達すると報告している41).また,その後の解析において,がん細胞上のB7-H1(PD-L1)が,PD-1以外の分子を介してT細胞にアポトーシスを誘導することにより,抗腫瘍免疫応答を阻害していると報告している42).しかし,現在までに,PD-L1と結合してアポトーシスを誘導する受容体は報告されておらず,その分子メカニズムは不明である.
9. ヒトがん組織におけるPD-L1,PD-L2の発現
がん細胞株に加え,ヒトがん組織においてもPD-L1やPD-L2の発現が認められた.また,がん組織におけるPD-L1やPD-L2の発現が,がん患者の生命予後と逆相関することが複数のがん腫において見いだされたことから,PD-1が実際のがん患者においても抗腫瘍免疫応答を減弱していることが強く示唆された.Thompsonらは,腎がんの術後3年生存率が,摘出がん組織におけるPD-L1の発現が強かった患者群では63.2%,発現が弱かった患者群では88.4%であったと報告している43).がん組織におけるPD-L1の発現強度と生命予後の逆相関は,腎がん以外にも,卵巣がん,胃がん,食道がん,膵がん,メラノーマ等においても認められている2,44).このことは,これらのがん腫において,(1)がん細胞を免疫システムが異物と認識して,がん細胞に特異的なTリンパ球が反応していること,(2)がん細胞がPD-1リガンドの発現を獲得することがあること,および(3)がん細胞上のPD-1リガンドが,がん細胞に特異的な細胞傷害性T細胞の活性を抑制することを示唆する.すなわち,がん細胞にPD-1リガンドが発現していない症例では,がん細胞に特異的なT細胞が適切にがん細胞を攻撃して死滅させているため,予後がよかったものと推測される.一方,がん細胞にPD-1リガンドが発現している症例では,がん細胞に特異的なT細胞は存在するものの,がん細胞を攻撃できないため,がん細胞が増殖を続け,予後が悪かったものと推測される.この推論およびマウスにおける治療結果からは,PD-1を阻害するだけで,がん細胞特異的T細胞が本来の機能を発揮し,がん細胞を効果的に排除できると考えられる.
以上のような前臨床研究に基づいて,がん免疫賦活薬としてPD-1阻害抗体が開発され,米国で2006年から,日本では2009年から臨床試験が開始された.2010年に米国における第1相臨床試験の結果として,39例のうち1例で完全寛解が,2例で部分寛解が認められたと報告され,その効果に大きな関心が寄せられた45).2012年にTopalianらによって結果が報告されたより大規模な臨床試験では,悪性黒色腫94例のうち26例(28%),腎細胞がん33例のうち9例(27%)および肺非小細胞がん76例のうち14例(18%)に奏効(完全寛解あるいは部分寛解)が認められたが,ホルモン療法抵抗性の前立腺がん(17例)と大腸がん(19例)では,奏効例は認められなかった46).本試験においては,治療開始前のがん組織にPD-L1が発現している症例では高い奏効率(25例中9例,36%)が得られたが,PD-L1を発現していない症例では奏効例が認められなかった(17例中0例)と報告されている.その後の報告の中には,治療効果と相関しないとするものもあることから47),治療開始前のがん組織におけるPD-L1の発現の有無を治療効果の予測に利用することについては,慎重な検討が必要である.具体的な延命効果としては,第Ⅲ・Ⅳ病期の切除不能なメラノーマ患者において,1年生存率がPD-1阻害抗体治療群で72.9%,化学療法ダカルバジン投与群で42.1%であったという報告がある48).また,ホジキンリンパ腫においては,87%というきわめて高い奏効率が報告される等,一部のがん腫においては特に高い治療効果が認められている49).さらに,CTLA-4阻害抗体とPD-1阻害抗体の併用がメラノーマの治療において劇的な相乗効果を示し,52例中16例において,治療開始後12週間以内に80%以上の腫瘍縮小効果が認められたと報告されている50).PD-L1阻害抗体についても第1相臨床試験の結果が報告されており,PD-1阻害抗体と比較して少し奏効率が低い傾向はあるが,同様に好ましい治療効果が得られている51).
以上のような臨床試験の結果を受けて,日本で2014年7月に,米国では2014年9月に切除不能なメラノーマに対する使用が認可された.また,2015年3月には米国において,非小細胞性肺がんに対しての使用も認可されている.
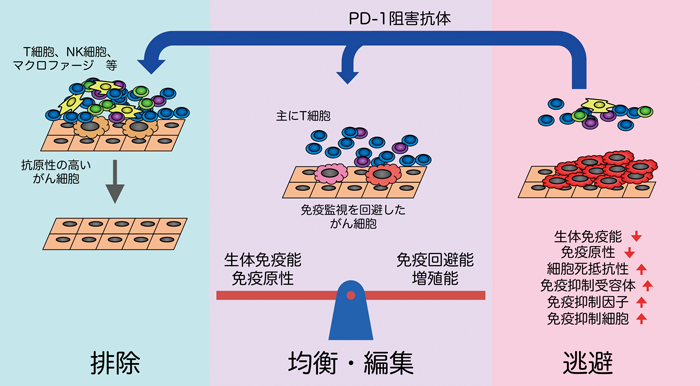
がん細胞を免疫システムが異物と認識して排除するかどうかは免疫学者にとって大きな謎であり,古くから議論が繰り返されてきた.Burnetは,1950年代から70年代にかけて,immunosurveillance(免疫監視)という概念を提唱した.すなわち,脊椎動物をはじめとした長寿の動物では,体細胞に遺伝子の変異が蓄積してがん化することは一般的であるため,がん化した細胞を排除する機構を獲得することが進化学上の必然であり,その機構は免疫系が担っているに違いないという仮説を提唱した.その後,がん細胞に対する免疫監視の存在を支持する実験結果が得られず,むしろ免疫応答に伴う炎症によりがん化が促進されるという実験結果の方が多く得られる等,長らくがんと免疫の関係は混沌としていた.近年,免疫監視の概念に修正が加えられたimmuno-editing(免疫編集)という概念が提唱されているが,PD-1阻害抗体によるがん治療の結果は,正しくimmuno-editingの概念を支持している(図6)5,6).本概念では,がんと免疫の関係は,排除(elimination),均衡状態(equilibrium),逃避(escape)の3段階に分けられる.まず,がん細胞に対する免疫監視は確かに存在し,がん化した細胞は抗腫瘍免疫応答によって排除されるが,一部のがん細胞が排除を逃れて長期に生存して均衡状態に至ることがある.均衡状態では,がん細胞が遺伝子変異の蓄積等により増殖能と免疫逃避能を向上させるのに対して,獲得免疫系が何とか抑え込んでいる,いわばdormantな状態である.ここで決定的な遺伝子変異の獲得あるいは環境の変化により均衡状態が崩れると,免疫監視から逃避して,がん細胞が異常増殖して腫瘍が形成される.
均衡状態において,がん細胞が獲得免疫系によりdormantな状態にとどめおかれている証拠としては,野生型マウスに化学発がん物質を投与して腫瘍が形成されなかった個体において,T細胞除去抗体等により免疫不全を誘導すると,腫瘍が形成されたという報告等がある52).ヒトにおいても,治療によりがんが寛解した患者をドナーとした臓器移植において,レシピエントがドナー由来のがんを発症したという報告が多数ある.顕著な例としては,16年前にメラノーマを発症し,外科切除により寛解していた患者から腎臓の提供を受けたレシピエントが,ドナー由来のメラノーマを発症したという例がある53).ドナーの中で16年間にわたってdormancyを保っていたメラノーマが,拒絶を抑えるための免疫抑制剤により均衡が崩れ,いわば逃避状態となって腫瘍を形成したと考えられる.
均衡状態から逃避に移行する要因としては,免疫抑制剤の使用に加え,がん細胞における抗原性の変化,細胞死に対する抵抗性の獲得,免疫抑制能の獲得等が考えられる.抗原性の変化については,抗原そのものの変異や発現低下に加え,MHCの発現低下や抗原提示に関わる遺伝子の異常等の関与が報告されている.細胞死に対する抵抗性の獲得については,Bcl-xl等のアポトーシス抵抗因子の発現,Fas等の細胞死誘導受容体の変異等の関与が報告されている.免疫抑制能の獲得については,PD-L1等の抑制性リガンドの発現,Treg(regulatory T cell)やMDSC(myeloid-derived suppressor cell)の誘引,TGF-β,IDO,アルギナーゼ等の抑制性因子の発現等の関与が報告されている5,6).
腫瘍として発見されるまでに巨大化したがん細胞は,何らかの手段を用いて免疫監視から逃避していると考えられるが,どのような手段を獲得したかは,がん細胞がどのような免疫監視の圧力を受けていたかによって異なる.すなわち,免疫環境によってがん細胞の特性が変化することを意味し,immuno-editingと呼ばれる.これまでにimmuno-editingの存在を支持する実験結果も複数得られている.Shankaranらは,免疫健常マウスにおいて化学発がん物質により発生した腫瘍は,免疫健常マウスに接種するとすべてが生着するが,免疫不全マウスにおいて化学発がん物質により発生した腫瘍は,免疫健常マウスに接種すると約半数が生着しないと報告している54).免疫不全マウスでは,免疫監視の圧力がないためimmuno-editingを十分に受けておらず,細胞増殖に関する遺伝子変異は十分に蓄積しているものの,免疫逃避に関する遺伝子変異を十分に獲得していないために,免疫健常マウスに移植した際に,免疫監視によって排除されてしまったと考えられる.
前述のとおり,ヒトがん組織におけるPD-L1あるいはPD-L2の発現が生命予後と逆相関するとともに,議論の余地はまだあるものの,がん組織におけるPD-L1の発現がPD-1阻害抗体の効果と相関することが複数報告されていることは,ヒトに発生するがんにおいて,PD-1システムを用いてがん免疫応答を減弱することが,均衡状態から抜け出し,免疫監視から逃避する上で,決定的な役割を果たしていることを示唆する.また,immuno-editingという概念を強く支持すると考えられる.もっとも,immuno-editingの概念に従うと,PD-1阻害抗体によってがん細胞が完全に排除されたと思っても,実際には新たな均衡状態が始まるだけともいえる.すなわち,PD-1リガンド発現によるPD-1システム悪用とは別の手法を用いて免疫監視から逃避し,腫瘍を再形成する可能性がある.
従来の化学療法では,投与した直後には腫瘍縮小効果が認められるが,効果が持続しないために,次々と薬剤を切り替えるということがよく行われている.このとき,がん細胞が,いわばchemo-editingを受け,各薬剤に抵抗性のがん細胞が選択されることにより,細胞増殖に関わる経路が次々に強化されている可能性がある.病原微生物の多剤耐性菌とも通じるものがあるが,染色体不安定性を特色とするがん細胞の治療にあたっては,いたずらに均衡状態を長引かせるのではなく,寄与率の高い経路を標的とすることで,確実に排除を惹起する,あるいは強力にdormantな状態に閉じ込めることがきわめて重要だと考えられる.
がん免疫は一種の自己免疫であると述べたが,がん免疫が実際に自己抗原を標的としているかについては,明確な証明はなかった.近年,PD-1阻害抗体が高い確率で奏功したことから,効果的ながん免疫の標的について,より直接的な検証が可能となった.Rizviらは,非小細胞性肺がんを用いて全エクソンの塩基配列解析を行ったところ,がん組織においてアミノ酸置換を伴う変異の数が多いほど,PD-1阻害抗体の奏効率が高かったと報告している55).このことは,PD-1阻害抗体による腫瘍退縮効果は,完全なる自己抗原ではなく,がん組織において生み出された変異抗原(ネオ抗原)を認識するTリンパ球の活性化によることを示唆する.ネオ抗原を認識するTリンパ球は,胸腺における負の選択を受けない点において自己反応性T細胞と異なるが,末梢において副刺激分子を有さない実質細胞に抗原提示を受ける点においては自己反応性T細胞と共通である.
がん細胞に生じる遺伝子変異のうち,無秩序な増殖能の獲得に直接関係する変異はドライバー変異,直接には関係しない変異はパッセンジャー変異と呼ばれて区別されている.ドライバー変異を含む部分ペプチドがネオ抗原として機能すれば,同一のドライバー変異を有するがん腫に共通の標的となることから,免疫応答の評価やワクチンに利用できると期待されて,精力的に検討されてきた.近年,メラノーマにおいて標的とされるネオ抗原のほとんどはパッセンジャー変異であることが明らかとされている6).また,がんゲノム解析において同定されたアミノ酸変異を伴う遺伝子変異について,MHCとの親和性を基に変異アミノ酸を含む部分ペプチドがネオ抗原として提示される可能性を予測した解析では,ランダムなアミノ酸変異と比較してネオ抗原として提示される確率が有意に低いとする実験結果が報告された56).抗原性を高めない遺伝子変異が選択された結果と考えられるが,immuno-editingの概念にまさしく合致する.PD-1阻害抗体は,ネオ抗原の特性を問わずに効果を示すことから,きわめて効果的といえる.今後,さまざまながん腫において,PD-1阻害時および非阻害時にT細胞の標的とされる抗原が,個々の症例で異なるのか,あるいは何らかの主要な抗原が存在するのかが明らかにされると期待される.造血系は細胞増殖が盛んであることから,一般的に化学療法は免疫療法との併用に適していないと考えられているが,chemo-editingにより抗原性の高い変異が導入されて,腫瘍免疫への感受性が向上する可能性も考えられる.現在,化学療法を含むさまざまな併用療法が検討されているが,併用による相乗効果と弊害について,新たな知見が得られると期待される.
前述のとおりPD-1欠損マウスが自己免疫疾患を自然発症することから,PD-1阻害抗体によるがん治療の副作用として,自己免疫疾患の発症が懸念された.実際,これまでに,間質性肺炎,腸炎,発疹,Ⅰ型糖尿病,心筋炎,腎障害,肝機能障害,甲状腺機能低下症,尿閉,関節炎,重症筋無力症等の発症が報告されている45–51,57–59).CTLA-4抗体による副作用と比較すると発症頻度と重症度ともに軽度ではあるが60,61),重症化して死亡した例も報告されているとともに,適応がん腫が拡大することにより,これまでに報告されているものとは異なる自己免疫疾患が発症する可能性もあるため,厳重な注意が必要である.
興味深いことに,上記症状のほとんどが,PD-1欠損マウスにおいても認められる.一部の症状については,NODやMRL系統あるいはLAG-3やFcγRIIBの同時欠損下でのみ各疾患を発症することから,特殊な条件下においてのみ観察される特殊な現象と捉える意見もあったが,数百人規模の臨床試験においてこれらの症状が観察されたことから,ヒトの遺伝的多様性の前では決して特殊なものでは無いと判断される.逆に,実際の生命現象を理解するためには様々な遺伝背景のモデルマウスを用いて解析することが重要であると言える.NOD.H2b-PD-1欠損マウスを用いた連鎖解析の結果が示すとおり,多遺伝子疾患では,複数の疾患感受性遺伝子を併せ持つ患者では症状が激しくなるとともに,疾患感受性遺伝子を少ししか持たなくても,低頻度ながら発症する可能性がある.したがって,PD-1阻害抗体の副作用を予測する上で,自己免疫素因の有無は重要な要素ではあるが,完全ではないため,副作用に対しては十分な注意が必要である.
自己免疫,がん免疫に加え,PD-1は感染免疫にも大きく関与する.PD-1欠損マウスにアデノウイルスを感染させると,野生型マウスよりも速やかにウイルスが排除されたが,感染細胞の傷害による激しい組織破壊を伴ったことから,PD-1による感染免疫応答の抑制は,急激な感染免疫応答による組織破壊の防止を目的としていると考えられた62).その後,Barberらによって,慢性ウイルス感染症において持続的な抗原刺激により機能不全の状態に陥ったウイルス特異的T細胞にPD-1が発現しており,PD-1が機能不全状態の維持に関与していることが見いだされた11,12).また,慢性に感染状態が維持されるウイルスや結核菌の感染においても,PD-1あるいはPD-L1欠損マウスが,激しい炎症を惹起して感染後早期に死亡することが報告されている63).ヒトにおいても,HCV,HBV,HIV等,慢性に感染状態が維持されるウイルスの感染において,各ウイルス特異的なT細胞上にPD-1が発現して機能を減弱していることが報告されている64,65).一方,ウイルス,細菌,寄生虫等の感染により,感染細胞や抗原提示細胞上にPD-1リガンドの発現が誘導され,これらの微生物に対する免疫応答が減弱されることも明らかとなっている2,66).進化学上,病原微生物が宿主の免疫応答によってeditされていると考えられるが,PD-1やPD-L1の発現誘導も,宿主の免疫応答から逃避するために病原微生物が獲得した戦略の一つかもしれない.
すでにヒトHCV患者に対するPD-1阻害抗体の臨床試験が米国において開始されているが,急激な免疫応答による劇症肝炎の発症には十分な注意が必要と思われる.また,がんの治療においても,何らかの慢性感染ウイルスに罹患している患者においては,十分な注意が必要と思われる.
がんの治療においてPD-1阻害抗体が劇的な治療効果を示したことから,また一部の患者が重篤な自己免疫疾患を副作用として発症したことから,ヒト生体内において,PD-1が実際に自己やがん細胞に対する免疫応答を抑制することにより,生体の恒常性を維持していることが明らかとなった.しかし,劇的な効果とはいうものの,その奏功率は20~30%であり,半分以上の症例では奏功しない.前述のとおり,PD-1は遺伝背景や環境によって異なる分子と協調的に自己免疫疾患の発症を制御すると考えられているが,おそらくがん免疫においても同様に,他の分子と協調的に機能しているものと思われる.今後,さまざまな環境において,PD-1がどのような分子と協調的に機能しているかが解明されれば,免疫寛容の成立・維持に関わるネットワークシステムの全貌が明らかになるとともに,より効果的な腫瘍免疫療法の開発につながると期待される.また,本稿ではがんの治療を中心に紹介したが,PD-1の機能を阻害することにより,感染症やアレルギー疾患の治療にも役立つ可能性がある.また,PD-1の機能を増強することができれば,自己免疫疾患や移植片拒絶,劇症型感染症の治療につながると期待される.
引用文献References
1) Ishida, Y., Agata, Y., Shibahara, K., & Honjo, T. (1992) EMBO J., 11, 3887–3895.
2) Okazaki, T. & Honjo, T. (2007) Int. Immunol., 19, 813–824.
3) Okazaki, T., Chikuma, S., Iwai, Y., Fagarasan, S., & Honjo, T. (2013) Nat. Immunol., 14, 1212–1218.
4) Chen, L. & Flies, D.B. (2013) Nat. Rev. Immunol., 13, 227–242.
5) Schreiber, R.D., Old, L.J., & Smyth, M.J. (2011) Science, 331, 1565–1570.
6) Schumacher, T.N. & Schreiber, R.D. (2015) Science, 348, 69–74.
7) Nishimura, H., Minato, N., Nakano, T., & Honjo, T. (1998) Int. Immunol., 10, 1563–1572.
8) Nishimura, H., Nose, M., Hiai, H., Minato, N., & Honjo, T. (1999) Immunity, 11, 141–151.
9) Okazaki, T., Maeda, A., Nishimura, H., Kurosaki, T., & Honjo, T. (2001) Proc. Natl. Acad. Sci. USA, 98, 13866–13871.
10) Kawamoto, S., Tran, T.H., Maruya, M., Suzuki, K., Doi, Y., Tsutsui, Y., Kato, L.M., & Fagarasan, S. (2012) Science, 336, 485–489.
11) Barber, D.L., Wherry, E.J., Masopust, D., Zhu, B., Allison, J.P., Sharpe, A.H., Freeman, G.J., & Ahmed, R. (2006) Nature, 439, 682–687.
12) Okazaki, T. & Honjo, T. (2006) Cell, 124, 459–461.
13) Shimatani, K., Nakashima, Y., Hattori, M., Hamazaki, Y., & Minato, N. (2009) Proc. Natl. Acad. Sci. USA, 106, 15807–15812.
14) Freeman, G.J., Long, A.J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L.J., Malenkovich, N., Okazaki, T., Byrne, M.C., Horton, H.F., Fouser, L., Carter, L., Ling, V., Bowman, M.R., Carreno, B.M., Collins, M., Wood, C.R., & Honjo, T. (2000) J. Exp. Med., 192, 1027–1034.
15) Latchman, Y., Wood, C.R., Chernova, T., Chaudhary, D., Borde, M., Chernova, I., Iwai, Y., Long, A.J., Brown, J.A., Nunes, R., Greenfield, E.A., Bourque, K., Boussiotis, V.A., Carter, L.L., Carreno, B.M., Malenkovich, N., Nishimura, H., Okazaki, T., Honjo, T., Sharpe, A.H., & Freeman, G.J. (2001) Nat. Immunol., 2, 261–268.
16) Ishida, M., Iwai, Y., Tanaka, Y., Okazaki, T., Freeman, G.J., Minato, N., & Honjo, T. (2002) Immunol. Lett., 84, 57–62.
17) Yamazaki, T., Akiba, H., Iwai, H., Matsuda, H., Aoki, M., Tanno, Y., Shin, T., Tsuchiya, H., Pardoll, D.M., Okumura, K., Azuma, M., & Yagita, H. (2002) J. Immunol., 169, 5538–5545.
18) Guleria, I., Khosroshahi, A., Ansari, M.J., Habicht, A., Azuma, M., Yagita, H., Noelle, R.J., Coyle, A., Mellor, A.L., Khoury, S.J., & Sayegh, M.H. (2005) J. Exp. Med., 202, 231–237.
19) Hori, J., Wang, M., Miyashita, M., Tanemoto, K., Takahashi, H., Takemori, T., Okumura, K., Yagita, H., & Azuma, M. (2006) J. Immunol., 177, 5928–5935.
20) Brownlie, R.J. & Zamoyska, R. (2013) Nat. Rev. Immunol., 13, 257–269.
21) Sheppard, K.A., Fitz, L.J., Lee, J.M., Benander, C., George, J.A., Wooters, J., Qiu, Y., Jussif, J.M., Carter, L.L., Wood, C.R., & Chaudhary, D. (2004) FEBS Lett., 574, 37–41.
22) Yokosuka, T., Takamatsu, M., Kobayashi-Imanishi, W., Hashimoto-Tane, A., Azuma, M., & Saito, T. (2012) J. Exp. Med., 209, 1201–1217.
23) Yokosuka, T., Kobayashi, W., Takamatsu, M., Sakata-Sogawa, K., Zeng, H., Hashimoto-Tane, A., Yagita, H., Tokunaga, M., & Saito, T. (2010) Immunity, 33, 326–339.
24) Nishimura, H., Okazaki, T., Tanaka, Y., Nakatani, K., Hara, M., Matsumori, A., Sasayama, S., Mizoguchi, A., Hiai, H., Minato, N., & Honjo, T. (2001) Science, 291, 319–322.
25) Okazaki, T., Otaka, Y., Wang, J., Hiai, H., Takai, T., Ravetch, J.V., & Honjo, T. (2005) J. Exp. Med., 202, 1643–1648.
26) Wang, J., Okazaki, I.M., Yoshida, T., Chikuma, S., Kato, Y., Nakaki, F., Hiai, H., Honjo, T., & Okazaki, T. (2010) Int. Immunol., 22, 443–452.
27) Wang, J., Yoshida, T., Nakaki, F., Hiai, H., Okazaki, T., & Honjo, T. (2005) Proc. Natl. Acad. Sci. USA, 102, 11823–11828.
28) Okazaki, T., Tanaka, Y., Nishio, R., Mitsuiye, T., Mizoguchi, A., Wang, J., Ishida, M., Hiai, H., Matsumori, A., Minato, N., & Honjo, T. (2003) Nat. Med., 9, 1477–1483.
29) Chikuma, S., Terawaki, S., Hayashi, T., Nabeshima, R., Yoshida, T., Shibayama, S., Okazaki, T., & Honjo, T. (2009) J. Immunol., 182, 6682–6689.
30) Francisco, L.M., Salinas, V.H., Brown, K.E., Vanguri, V.K., Freeman, G.J., Kuchroo, V.K., & Sharpe, A.H. (2009) J. Exp. Med., 206, 3015–3029.
31) Wicker, L.S., Todd, J.A., & Peterson, L.B. (1995) Annu. Rev. Immunol., 13, 179–200.
32) Yoshida, T., Jiang, F., Honjo, T., & Okazaki, T. (2008) Proc. Natl. Acad. Sci. USA, 105, 3533–3538.
33) Jiang, F., Yoshida, T., Nakaki, F., Terawaki, S., Chikuma, S., Kato, Y., Okazaki, I.M., Honjo, T., & Okazaki, T. (2009) Int. Immunol., 21, 499–509.
34) Honjo, T., Kinoshita, K., & Muramatsu, M. (2002) Annu. Rev. Immunol., 20, 165–196.
35) Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y., & Honjo, T. (2000) Cell, 102, 553–563.
36) Okazaki, T., Okazaki, I.M., Wang, J., Sugiura, D., Nakaki, F., Yoshida, T., Kato, Y., Fagarasan, S., Muramatsu, M., Eto, T., Hioki, K., & Honjo, T. (2011) J. Exp. Med., 208, 395–407.
37) Woo, S.R., Turnis, M.E., Goldberg, M.V., Bankoti, J., Selby, M., Nirschl, C.J., Bettini, M.L., Gravano, D.M., Vogel, P., Liu, C.L., Tangsombatvisit, S., Grosso, J.F., Netto, G., Smeltzer, M.P., Chaux, A., Utz, P.J., Workman, C.J., Pardoll, D.M., Korman, A.J., Drake, C.G., & Vignali, D.A. (2012) Cancer Res., 72, 917–927.
38) Kido, M., Watanabe, N., Okazaki, T., Akamatsu, T., Tanaka, J., Saga, K., Nishio, A., Honjo, T., & Chiba, T. (2008) Gastroenterology, 135, 1333–1343.
39) Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T., & Minato, N. (2002) Proc. Natl. Acad. Sci. USA, 99, 12293–12297.
40) Iwai, Y., Terawaki, S., & Honjo, T. (2005) Int. Immunol., 17, 133–144.
41) Dong, H., Zhu, G., Tamada, K., & Chen, L. (1999) Nat. Med., 5, 1365–1369.
42) Dong, H., Strome, S.E., Salomao, D.R., Tamura, H., Hirano, F., Flies, D.B., Roche, P.C., Lu, J., Zhu, G., Tamada, K., Lennon, V.A., Celis, E., & Chen, L. (2002) Nat. Med., 8, 793–800.
43) Thompson, R.H., Gillett, M.D., Cheville, J.C., Lohse, C.M., Dong, H., Webster, W.S., Krejci, K.G., Lobo, J.R., Sengupta, S., Chen, L., Zincke, H., Blute, M.L., Strome, S.E., Leibovich, B.C., & Kwon, E.D. (2004) Proc. Natl. Acad. Sci. USA, 101, 17174–17179.
44) Hamanishi, J., Mandai, M., Iwasaki, M., Okazaki, T., Tanaka, Y., Yamaguchi, K., Higuchi, T., Yagi, H., Takakura, K., Minato, N., Honjo, T., & Fujii, S. (2007) Proc. Natl. Acad. Sci. USA, 104, 3360–3365.
45) Brahmer, J.R., Drake, C.G., Wollner, I., Powderly, J.D., Picus, J., Sharfman, W.H., Stankevich, E., Pons, A., Salay, T.M., McMiller, T.L., Gilson, M.M., Wang, C., Selby, M., Taube, J.M., Anders, R., Chen, L., Korman, A.J., Pardoll, D.M., Lowy, I., & Topalian, S.L. (2010) J. Clin. Oncol., 28, 3167–3175.
46) Topalian, S.L., Hodi, F.S., Brahmer, J.R., Gettinger, S.N., Smith, D.C., McDermott, D.F., Powderly, J.D., Carvajal, R.D., Sosman, J.A., Atkins, M.B., Leming, P.D., Spigel, D.R., Antonia, S.J., Horn, L., Drake, C.G., Pardoll, D.M., Chen, L., Sharfman, W.H., Anders, R.A., Taube, J.M., McMiller, T.L., Xu, H., Korman, A.J., Jure-Kunkel, M., Agrawal, S., McDonald, D., Kollia, G.D., Gupta, A., Wigginton, J.M., & Sznol, M. (2012) N. Engl. J. Med., 366, 2443–2454.
47) Brahmer, J., Reckamp, K.L., Baas, P., Crino, L., Eberhardt, W.E., Poddubskaya, E., Antonia, S., Pluzanski, A., Vokes, E.E., Holgado, E., Waterhouse, D., Ready, N., Gainor, J., Aren Frontera, O., Havel, L., Steins, M., Garassino, M.C., Aerts, J.G., Domine, M., Paz-Ares, L., Reck, M., Baudelet, C., Harbison, C.T., Lestini, B., & Spigel, D.R. (2015) N. Engl. J. Med., 373, 123–135.
48) Robert, C., Long, G.V., Brady, B., Dutriaux, C., Maio, M., Mortier, L., Hassel, J.C., Rutkowski, P., McNeil, C., Kalinka-Warzocha, E., Savage, K.J., Hernberg, M.M., Lebbe, C., Charles, J., Mihalcioiu, C., Chiarion-Sileni, V., Mauch, C., Cognetti, F., Arance, A., Schmidt, H., Schadendorf, D., Gogas, H., Lundgren-Eriksson, L., Horak, C., Sharkey, B., Waxman, I.M., Atkinson, V., & Ascierto, P.A. (2015) N. Engl. J. Med., 372, 320–330.
49) Ansell, S.M., Lesokhin, A.M., Borrello, I., Halwani, A., Scott, E.C., Gutierrez, M., Schuster, S.J., Millenson, M.M., Cattry, D., Freeman, G.J., Rodig, S.J., Chapuy, B., Ligon, A.H., Zhu, L., Grosso, J.F., Kim, S.Y., Timmerman, J.M., Shipp, M.A., & Armand, P. (2015) N. Engl. J. Med., 372, 311–319.
50) Wolchok, J.D., Kluger, H., Callahan, M.K., Postow, M.A., Rizvi, N.A., Lesokhin, A.M., Segal, N.H., Ariyan, C.E., Gordon, R.A., Reed, K., Burke, M.M., Caldwell, A., Kronenberg, S.A., Agunwamba, B.U., Zhang, X., Lowy, I., Inzunza, H.D., Feely, W., Horak, C.E., Hong, Q., Korman, A.J., Wigginton, J.M., Gupta, A., & Sznol, M. (2013) N. Engl. J. Med., 369, 122–133.
51) Brahmer, J.R., Tykodi, S.S., Chow, L.Q., Hwu, W.J., Topalian, S.L., Hwu, P., Drake, C.G., Camacho, L.H., Kauh, J., Odunsi, K., Pitot, H.C., Hamid, O., Bhatia, S., Martins, R., Eaton, K., Chen, S., Salay, T.M., Alaparthy, S., Grosso, J.F., Korman, A.J., Parker, S.M., Agrawal, S., Goldberg, S.M., Pardoll, D.M., Gupta, A., & Wigginton, J.M. (2012) N. Engl. J. Med., 366, 2455–2465.
52) Koebel, C.M., Vermi, W., Swann, J.B., Zerafa, N., Rodig, S.J., Old, L.J., Smyth, M.J., & Schreiber, R.D. (2007) Nature, 450, 903–907.
53) MacKie, R.M., Reid, R., & Junor, B. (2003) N. Engl. J. Med., 348, 567–568.
54) Shankaran, V., Ikeda, H., Bruce, A.T., White, J.M., Swanson, P.E., Old, L.J., & Schreiber, R.D. (2001) Nature, 410, 1107–1111.
55) Rizvi, N.A., Hellmann, M.D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J.J., Lee, W., Yuan, J., Wong, P., Ho, T.S., Miller, M.L., Rekhtman, N., Moreira, A.L., Ibrahim, F., Bruggeman, C., Gasmi, B., Zappasodi, R., Maeda, Y., Sander, C., Garon, E.B., Merghoub, T., Wolchok, J.D., Schumacher, T.N., & Chan, T.A. (2015) Science, 348, 124–128.
56) Rooney, M.S., Shukla, S.A., Wu, C.J., Getz, G., & Hacohen, N. (2015) Cell, 160, 48–61.
57) Hamid, O., Robert, C., Daud, A., Hodi, F.S., Hwu, W.J., Kefford, R., Wolchok, J.D., Hersey, P., Joseph, R.W., Weber, J.S., Dronca, R., Gangadhar, T.C., Patnaik, A., Zarour, H., Joshua, A.M., Gergich, K., Elassaiss-Schaap, J., Algazi, A., Mateus, C., Boasberg, P., Tumeh, P.C., Chmielowski, B., Ebbinghaus, S.W., Li, X.N., Kang, S.P., & Ribas, A. (2013) N. Engl. J. Med., 369, 134–144.
58) Hughes, J., Vudattu, N., Sznol, M., Gettinger, S., Kluger, H., Lupsa, B., & Herold, K.C. (2015) Diabetes Care, 38, e55–e57.
59) Laubli, H., Balmelli, C., Bossard, M., Pfister, O., Glatz, K., & Zippelius, A. (2015) J. Immunother. Cancer, 3, 11.
60) Hodi, F.S., O’Day, S.J., McDermott, D.F., Weber, R.W., Sosman, J.A., Haanen, J.B., Gonzalez, R., Robert, C., Schadendorf, D., Hassel, J.C., Akerley, W., van den Eertwegh, A.J., Lutzky, J., Lorigan, P., Vaubel, J.M., Linette, G.P., Hogg, D., Ottensmeier, C.H., Lebbe, C., Peschel, C., Quirt, I., Clark, J.I., Wolchok, J.D., Weber, J.S., Tian, J., Yellin, M.J., Nichol, G.M., Hoos, A., & Urba, W.J. (2010) N. Engl. J. Med., 363, 711–723.
61) Phan, G.Q., Yang, J.C., Sherry, R.M., Hwu, P., Topalian, S.L., Schwartzentruber, D.J., Restifo, N.P., Haworth, L.R., Seipp, C.A., Freezer, L.J., Morton, K.E., Mavroukakis, S.A., Duray, P.H., Steinberg, S.M., Allison, J.P., Davis, T.A., & Rosenberg, S.A. (2003) Proc. Natl. Acad. Sci. USA, 100, 8372–8377.
62) Iwai, Y., Terawaki, S., Ikegawa, M., Okazaki, T., & Honjo, T. (2003) J. Exp. Med., 198, 39–50.
63) Barber, D.L., Mayer-Barber, K.D., Feng, C.G., Sharpe, A.H., & Sher, A. (2011) J. Immunol., 186, 1598–1607.
64) Day, C.L., Kaufmann, D.E., Kiepiela, P., Brown, J.A., Moodley, E.S., Reddy, S., Mackey, E.W., Miller, J.D., Leslie, A.J., DePierres, C., Mncube, Z., Duraiswamy, J., Zhu, B., Eichbaum, Q., Altfeld, M., Wherry, E.J., Coovadia, H.M., Goulder, P.J., Klenerman, P., Ahmed, R., Freeman, G.J., & Walker, B.D. (2006) Nature, 443, 350–354.
65) Radziewicz, H., Ibegbu, C.C., Fernandez, M.L., Workowski, K.A., Obideen, K., Wehbi, M., Hanson, H.L., Steinberg, J.P., Masopust, D., Wherry, E.J., Altman, J.D., Rouse, B.T., Freeman, G.J., Ahmed, R., & Grakoui, A. (2007) J. Virol., 81, 2545–2553.
66) Kirchberger, S., Majdic, O., Steinberger, P., Bluml, S., Pfistershammer, K., Zlabinger, G., Deszcz, L., Kuechler, E., Knapp, W., & Stockl, J. (2005) J. Immunol., 175, 1145–1152.
著者紹介Author Profile
 岡崎 拓(おかざき たく)
岡崎 拓(おかざき たく)徳島大学疾患プロテオゲノム研究センターゲノム機能分野教授.博士(医学).
略歴1974年京都市で生まれる.99年京都大学医学部卒業.2003年同大学院医学研究科(本庶佑教授)修了.同年日本学術振興会特別研究員.同年同研究科分子生物学助手.04年同研究科21世紀COE特任助教授.08年より現職.
研究テーマと抱負自己免疫疾患の発症制御にかかわる分子ネットワークシステムの解明.免疫応答の人為的制御による自己免疫,がん,アレルギー等の治療.
ウェブサイトhttp://www.genome.tokushima-u.ac.jp/dir/
趣味美術鑑賞.スキー.お遍路.
 岡崎 一美(おかざき いるみ)
岡崎 一美(おかざき いるみ)徳島大学疾患プロテオゲノム研究センターゲノム機能分野准教授.博士(医学).
略歴1975年金沢市で生まれる.98年東京大学薬学部卒業.2000年同大学院薬学系研究科修士課程(入村達郎教授)修了.03年京都大学大学院医学研究科(本庶佑教授)博士課程修了.日本学術振興会特別研究員,京都大学大学院医学研究科助教を経て,08年より現所属.
研究テーマと抱負マウスを用いた自己免疫疾患のゲノム解析による,自己免疫疾患発症制御機構の解明.体細胞における遺伝子の編集・変化が自己免疫やがん免疫に与える影響の解明.
ウェブサイトhttp://www.genome.tokushima-u.ac.jp/dir/
趣味ピアノ.スキー.阿波おどり鑑賞(阿波おどりの時期に生命科学シンポジウムを開催しておりますので,興味のある方はご連絡下さい).