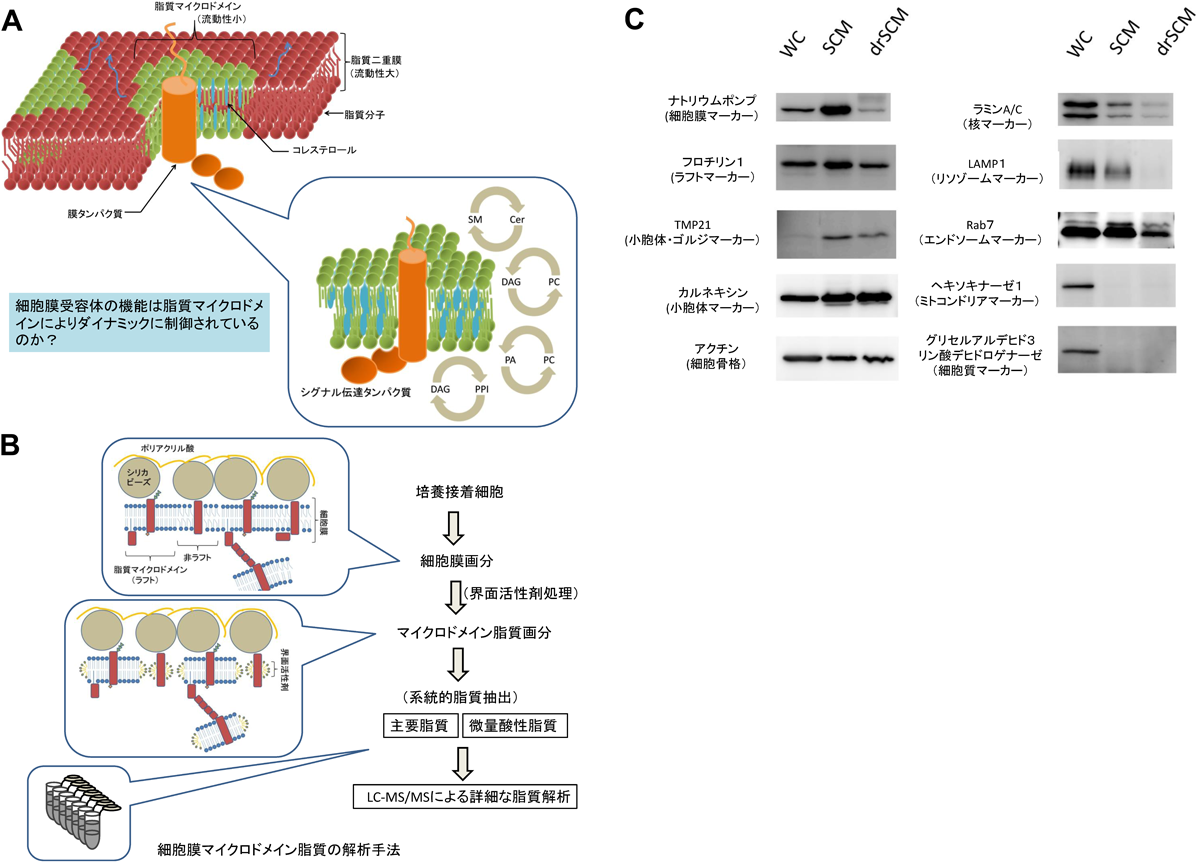
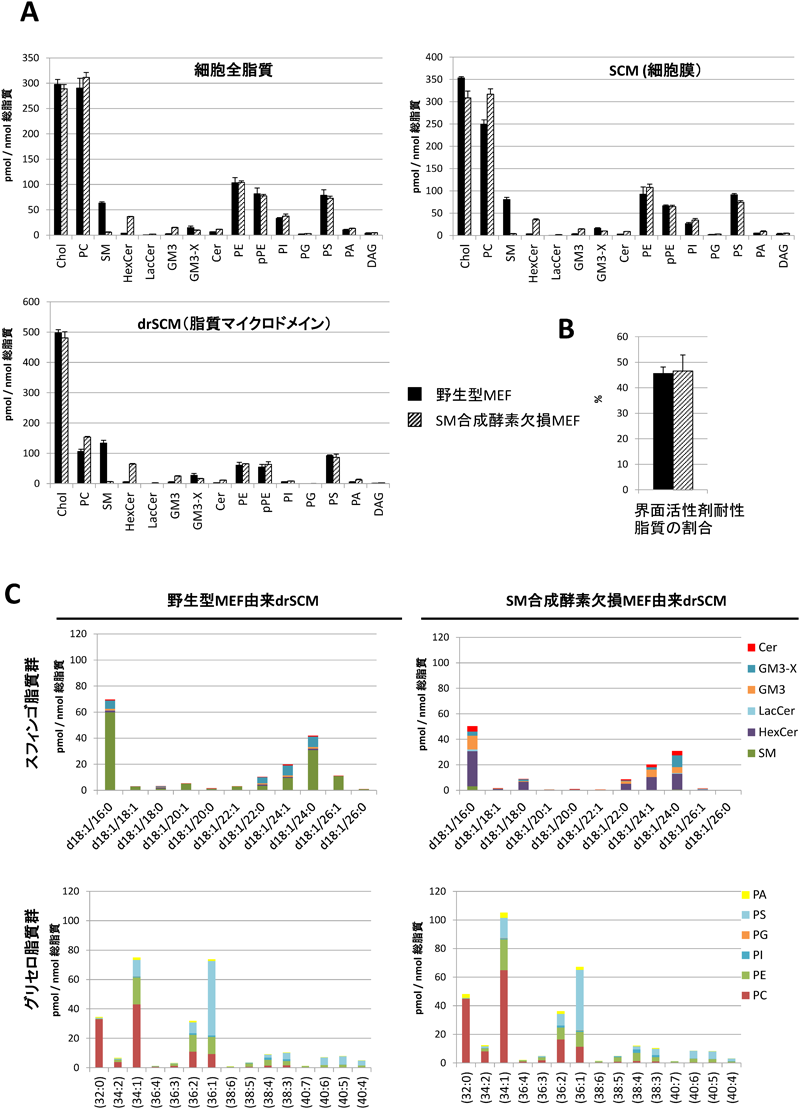
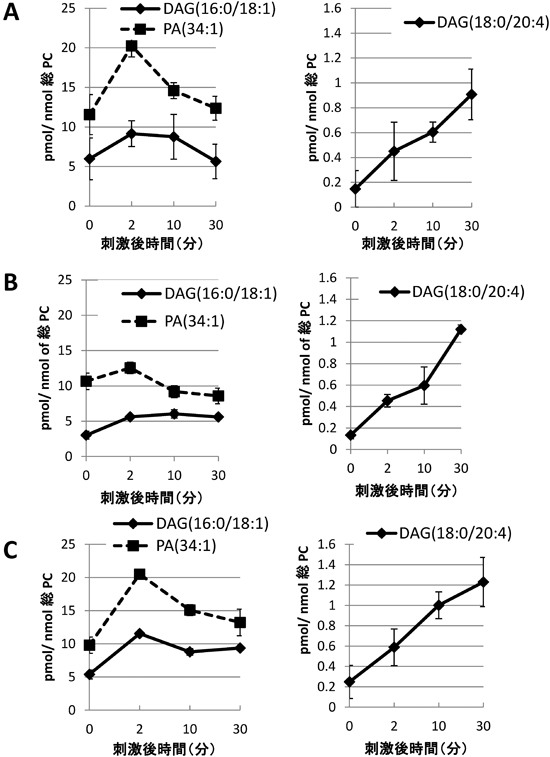
質量分析による細胞膜マイクロドメイン脂質の定量的解析法Quantitative analysis of lipids constructing plasma membrane microdomains by LC-MS
1 金沢医科大学医学部School of Medicine, Kanazawa Medical University ◇ 〒920-0293 石川県河北郡内灘町大学一丁目1番地Daigaku 1-1, Uchinada-machi, Kahoku-gun, Ishikawa 920-0293, Japan
2 金沢医科大学総合医学研究所先端医療研究領域加齢制御研究分野Division of Aging Research, Department of Advanced Medicine, Medical Research Institute, Kanazawa Medical University ◇ 〒920-0293 石川県河北郡内灘町大学一丁目1番地Daigaku 1-1, Uchinada-machi, Kahoku-gun, Ishikawa 920-0293, Japan
発行日:2015年12月25日Published: December 25, 2015