「ニュートリゲノミクス」は2000年代になって登場した言葉である.科研費のキーワードにも平成25年度から取り入れられ,筆者の専門である食品科学の分野では一定の市民権を得ているが,基礎生物学や医学,薬学の分野の方々にはあまりなじみがないかもしれない.お察しのとおり,栄養または栄養学を表すニュートリションとゲノミクスの複合造語であるが,実際には栄養素以外の物質も含む,食品を摂取した際の生体側の応答を(主として遺伝子発現の変化として)網羅的に解析すること,またはその学問分野を意味する.食品は薬品のような単一成分とは異なり,複雑な混合物である上に,生体側の応答も微少な変化であることが多い.そのような微少な変化を捉えるために,1995年に開発されたDNAマイクロアレイ1)によるトランスクリプトミクスが好んで用いられた.DNAマイクロアレイは,登場してからすでに20年が経ち,次世代シークエンサー(「次世代」という名称自体が現状に即さなくなってきているものの,通称として定着しているのでこの名称を使用する)の普及とあいまって,どちらかというと古い技術になりつつある.しかし,簡便で解析手法も確立されており,トランスクリプトミクスのツールとしては現在でもきわめて有用である.2015年7月に公開された映画「ターミネーター:新起動/ジェニシス」に登場する,“Old, but not obsolete.”(古いがポンコツではない)という印象的な台詞がDNAマイクロアレイにはぴったりあてはまる.今後は次世代シークエンサーによるトランスクリプトミクスが主流になってくるであろうことは筆者も否定しないが,DNAマイクロアレイでできることはまだたくさんあると考えている.
ご存知のように,DNAマイクロアレイはガラスなどの基板上に既知の塩基配列のオリゴDNAプローブを高密度に整列(アレイ)させたものである.筆者が使い始めた2005年当時は大きく分けて2種類のDNAマイクロアレイが使われていた.DNAマイクロアレイの開発者であるBrownらによる,スライドガラスにcDNAをスポットしたStanford型マイクロアレイと,光リソグラフィ技術を用いて基板上で直接DNA合成を行うAffymetrix型マイクロアレイ(以降GeneChip)である.前者はその後インクジェットプリンターの技術を導入することにより,プローブ密度が上がり,またロット間の製造誤差もかなり少なくなったが,当初はcDNA溶液をスポットするという製造原理に基づくロット間のばらつきや,ハイブリダイゼーションなどの工程に実験者の手技の巧拙が反映されやすいなどの問題があった.一方,後者は値段が高かったものの,プロトコールを含め人為的なエラーを徹底的に排除する思想で設計されていたため,実験者や実験日によらず比較的安定した結果が得られることから,筆者らはGeneChipを常用プラットフォームとして採用することとした.
ニュートリゲノミクスをはじめとする,遺伝子発現変動研究に用いられるDNAマイクロアレイデータ解析は,データの取得後,1)データの正規化,2)サンプル間クラスタリングによる遺伝子発現プロファイルの俯瞰,3)発現変動遺伝子の抽出,4)発現変動遺伝子セットの機能的特徴の解析,という流れで行われる.先にも述べたように,ニュートリゲノミクスは複雑なインプットに対するアウトプットの微少な差を検出することが要求される.そのため,筆者はこの分野で研究を始めるにあたり,実験デザインの洗練はもちろんのこと,解析も最適化する必要があると考えた.マイクロアレイは原理上バックグラウンドシグナルや非特異的結合などといったノイズがデータに含まれやすいので,より確実に発現変動遺伝子を選抜するためには,感度・特異度の高い解析手法が不可欠なのである.以下に筆者が通常ルーティンに行っている解析パイプラインの概要を記す.
1)DNAマイクロアレイデータの正規化
GeneChipは,プローブの設計にも特徴がある.Stanford型では50~60塩基長程度のcDNAプローブが配置されているのに対し,GeneChipでは1遺伝子につき25塩基長のプローブ配列を通常11か所設定している.しかも,それぞれのプローブには,25塩基のちょうど真ん中にあたる13番目の塩基にミスマッチ塩基を導入することによって,非特異的な結合を見積もるためのミスマッチプローブも設定している.完全に一致するものをパーフェクトマッチプローブといい,ミスマッチと合わせてプローブペアと呼ぶ.都合,1遺伝子に対して11組のプローブペアが設定されていることになる.これを,プローブセットという.このようなプローブデザインであることから,得られるデータも1遺伝子に対し22個のプローブの蛍光強度データ,ということになる.したがって,1遺伝子の発現強度は,これらの蛍光強度データを総合評価して算出される.これを正規化(summarization)といい,GeneChipの場合,バイオインフォマティクスの分野から正規化手法がいくつも提案されている.実験系の論文でこれまでよく使われてきたMicro Array Suite 5.0(MAS5.0)2),ここ数年Affymetrix社が提供している解析ソフトExpression Consoleにおけるデフォルトの正規化手法となっているRobust Multi-array Average(RMA)3)をはじめ,Factor Analysis for Robust Microarray Summarization(FARMS)4),Distribution Free Weighted method(DFW)5),robust radius-minimax(rmx)6)など多くの方法がある.一般論として,新しい手法はそれ以前に発表された方法に比べ何か優れた点があるため論文として採択されるわけなので,より「正しく」遺伝子発現プロファイルを反映する正規化手法であると考えられる.しかし,実際に自分のデータに適用した場合,新しい手法ほどよいとは限らないこともある.それは,これら正規化手法を提案する論文で用いられているテストデータは必ずしも実データではなく,人工的に作成したモデルデータである場合も少なくないからであると考えられる.そこで筆者は,1回のマイクロアレイ実験から得られるデータに対し,複数の代表的な正規化手法を適用してみて,その後サンプル間クラスタリングを行い,自分の仮説(すなわち,ある処理によって遺伝子発現が変動する)を最も支持するクラスタを形成する正規化手法を採用している.ある意味「都合のよい」正規化手法を選んでいることになるため,このようなやり方はいわゆるカンニングのようなものである,という批判はある.しかし,正規化手法に決定版はないので,どの正規化手法を採用するのも自由である.したがって,使用した正規化手法を明記すれば問題ないと考えている.筆者の経験上,最終的にFARMSかDFWを採用するケースが多い7–9).
2)サンプル間クラスタリングによる遺伝子発現プロファイルの俯瞰
正規化によって各遺伝子の発現強度を算出したら,次にどのサンプルとどのサンプルの遺伝子発現プロファイルが近いのかを調べつつ,全体を俯瞰するために,発現強度データに基づいてサンプル間クラスタリングを行う.DNAマイクロアレイデータ解析においてこのプロセスは欠かすことができない.というのは,重要な情報がクラスタリングの結果から得られるからである.バイオロジカルレプリケートどうしがクラスタを形成すれば,実験間の再現性がよい,ということになる.また,コントロール群と処理群のクラスタの分離の仕方をみれば,処理による遺伝子発現への影響の大きさをある程度把握できる.クラスタリングを行った段階で,実験自体がうまくいっているか,処理の条件は妥当であったか,などの判断が可能である.さらに,上述のようにGeneChipの場合は正規化手法によって結果が変わってくるため,どの正規化手法を採用するかはクラスタリングの結果をみる必要がある.階層的クラスタリングを試してみてうまくいっていないようにみえても,主成分分析を適用するとクラスタが分離することがある(後に実例を紹介する).簡単にあきらめてはいけないのである.
3)発現変動遺伝子の抽出
DNAマイクロアレイデータは数万の遺伝子発現強度からなる多次元のデータであるため,比較したい群が多群になると計算が複雑になってしまう.そのためか,筆者が知る限り多群に対応する使い勝手のよい解析手法がほとんどない.そこで,筆者は実験デザインの段階から,できるだけシンプルな二群間比較になるようにしている.ここでは,二群間比較による発現変動遺伝子抽出に絞って話を進める.発現変動遺伝子の抽出には,正規化によって得られた遺伝子の発現量情報をもとに算出した統計量によるランキングを用いる.二群間比較による発現変動遺伝子の抽出には,fold changeやt-統計量,あるいはその両者の組み合わせがよく用いられると思うが,いずれの方法も全体的に発現量の小さい遺伝子が上位にきやすい,という欠点がある.fold changeの場合,全体的に発現量が小さければ,当然分母となる群の発現量も小さくなり,値が大きくなりやすい.また,t-統計量の場合,統計量を算出する際に分母にばらつきの情報を含む項がくるが,これも全体的に発現量が小さければ必然的に分母が小さくなり,このような遺伝子が上位にきやすくなる.たとえ一定以下の発現量の遺伝子をフィルタリングで除外したとしても,閾値近辺の遺伝子が上位にきやすいという意味で,この傾向は変わらない.これら従来の統計量によるランキングの問題点を克服するために,KadotaらによってWeighted Average Difference(WAD)統計量が提案された10).WAD統計量は,log fold changeに発現量のダイナミックレンジを加味した重みづけを行い,「発現量が大きく変動した」遺伝子が上位にくるように工夫された統計量である.また,Breitlingらは,2群のサンプル間の総当たりで比をとり,そのランキングの相乗平均を統計量として用いるRank products統計量を提案している11).Rank productsもWADとは異なる原理に基づくものの,やはり「発現量が大きく変動した」遺伝子が上位にくる傾向が強く,WADと比較的近いランキングを返す.さらに,Kadotaらは,複数ある正規化手法と二群間比較による発現変動遺伝子ランキング手法の最適な組み合わせも提案している12).そのガイドラインによれば,RMA・FARMS・DFWはRank productsと相性がよく,WADは正規化手法によらず安定して感度・特異度の面で優れた結果を返すことがわかっている.筆者もこのガイドラインに従い,GeneChipで正規化手法としてMAS5.0を用いる場合(1群のサンプル数が2以下)やGeneChip以外のプラットフォーム(Agilent・Illumina・GE等)を用いる場合はWAD,それ以外は通常Rank productsを用いている.
読者の中には,DNAマイクロアレイの結果とリアルタイムPCRの結果が一致しないことに困った経験がおありの方も多いと思う.両者が一致しない理由は,マイクロアレイに搭載されているプローブが認識する塩基配列とリアルタイムPCRで増幅される塩基配列が必ずしも同じ部位ではないこと,リアルタイムPCRの標準化に使用される内在性コントロールの選択が不適切で,コントロール自体が発現変動してしまっているケースがあることなど,いくつかの理由が考えられる.しかしいずれにしろ,両者の結果が一致しないとどちらが正しいのか,という気持ち悪さが残る.一方,両者の結果が一致する場合は,ほぼ間違いなく発現変動していると考えてよいだろう.上述の正規化手法と発現変動遺伝子のランキング法の組み合わせガイドラインは,組み合わせを評価する際に,「DNAマイクロアレイの結果とリアルタイムPCRの結果の一致度が高いこと」を基準の一つにしている.実際にこのガイドラインに従って発現変動遺伝子を抽出すると,リアルタイムPCRで検証したときに,両者の結果が一致しない,いわゆる「はずれ」の遺伝子をつかむ確率が低くなることを実感している.
なお,筆者は上記1)~3)のプロセスを,統計解析言語環境R13)を用い,Bioconductor(http://www.bioconductor.org)14)から必要なパッケージを追加して行っている.RはDNAマイクロアレイ解析のみならず非常に応用範囲の広い高機能なソフトである上に無料なので,ぜひお試しいただきたい.筆者のように実験系からバイオインフォマティクスに足を踏み入れた人でも理解しやすく,科学技術振興機構バイオサイエンスデータベースセンター主催の統合データベース講習会や,統合TV(http://togotv.dbcls.jp/ja/)・R-Tips(http://cse.naro.affrc.go.jp/takezawa/r-tips/r.html)等をはじめとするウェブサイトの情報も充実している.筆者が用いている手法はすべて東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニットの門田幸二先生によるウェブサイト「(Rで)マイクロアレイデータ解析(http://www.iu.a.u-tokyo.ac.jp/~kadota/r.html)」に網羅されている.
4)発現変動遺伝子セットの機能的特徴の解析
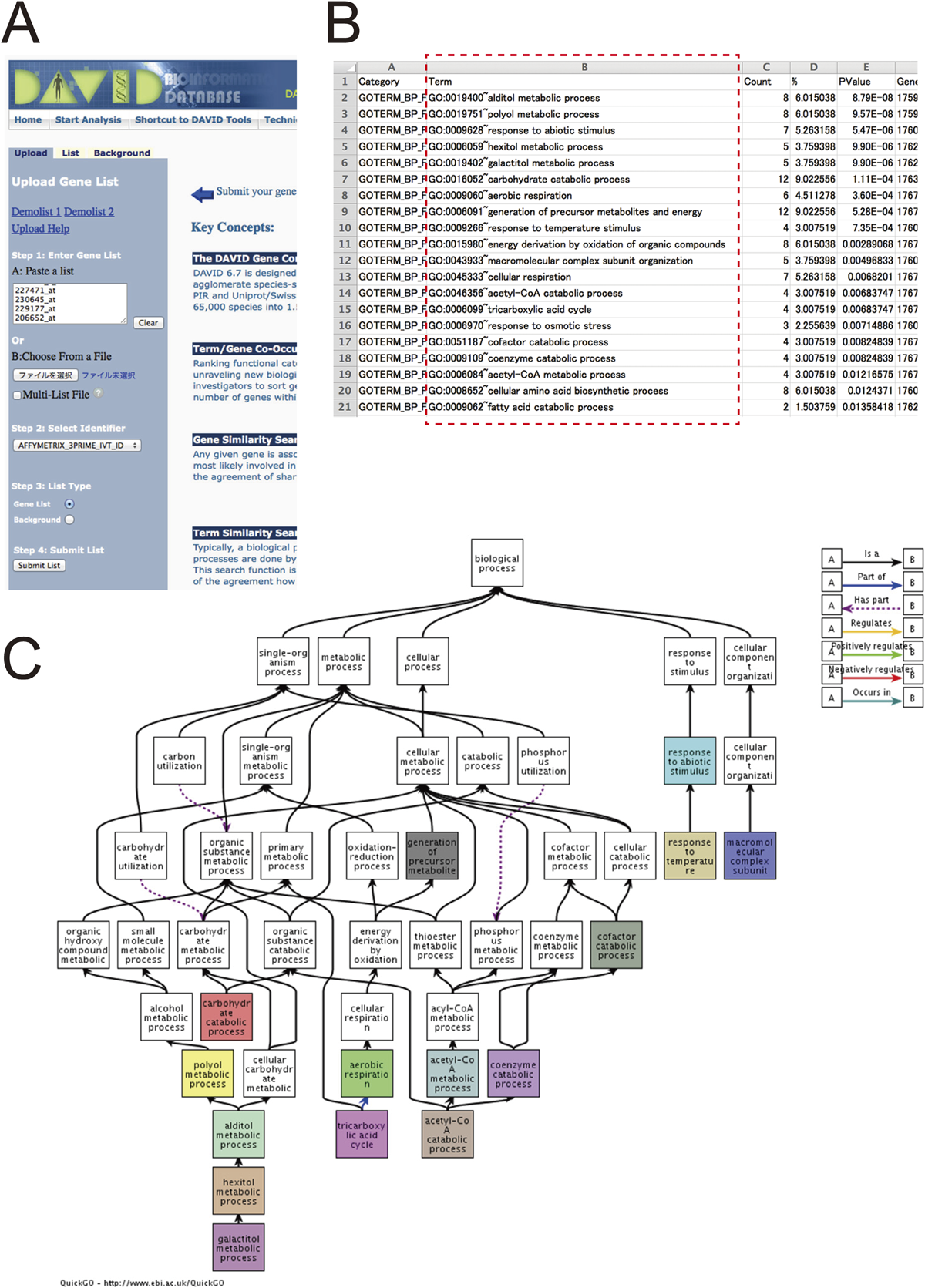
一定の条件を満たす発現変動遺伝子のセットが得られたら,それら遺伝子群にどういう機能的特徴があるか,というのは誰もが知りたいことであろう.ある処理に応答して,特定の機能を持った遺伝子の発現が変化するのであれば,得られた発現変動遺伝子セット中にはバックグラウンド(すなわちDNAマイクロアレイに搭載されている全遺伝子)に比べ,特定の機能を持った遺伝子が濃縮されているはずである.このように,バックグラウンドの遺伝子セットに対し,そこから抽出したサブセットの中にどのような機能を持った遺伝子が濃縮されているかを解析することを,gene-annotation enrichment analysis(エンリッチメント解析)という.筆者らは,主としてGene Ontology(GO)のアノテーション情報を用いて,the Database for Annotation, Visualization and Integrated Discovery(DAVID;http://david.abcc.ncifcrf.gov/)15)およびQuickGO(http://www.ebi.ac.uk/QuickGO/)16)というウェブツールを用いている.DAVIDは入力ウィンドウに発現遺伝子のIDを入力し,入力したIDの種類(Affymetrix ID, Gene Symbol等)を指定してsubmitボタンをクリックするだけのシンプルなインタフェイスで(図1A)使いやすい.QuickGOはGOのブラウザであるが,GO term(遺伝子の機能を記述する,階層的に整理された言葉)どうしの関係を示すterm comparisonという機能が便利である.DAVIDでのエンリッチメント解析の結果,得られるのは発現変動遺伝子セット中に濃縮されたGO termのリスト(図1B)である.このGO termのリストをQuickGOに入力し,term comparisonを行うと,GO termの階層性を加味したチャートとして図示してくれる(図1C).
その他,発現変動遺伝子リストを入力するとそれらの発現を制御している上流の転写因子を予測してくれる,TFactS(http://www.tfacts.org)17)というウェブツールもある.入力対応遺伝子IDがGene SymbolまたはEntrez IDに限られること,基本的にヒトの遺伝子セットにしか対応していないなどまだ改善の余地はあるものの,有用なツールである.
ニュートリゲノミクスは栄養学・機能性食品科学の流れをくむ研究分野であるが,DNAマイクロアレイ解析の部分をとり出してみれば,遺伝子発現変動を解析する研究であればどの研究分野にも当然のことながら適用可能である.特に,感度・特異度を最適化した,正規化手法と相性のよい発現変動遺伝子の抽出法の組み合わせは,従来の方法(MAS5.0とfold changeの組み合わせ等)に比べ解像度が上がり,新しい発見につながるといった好結果を生んでいる.本稿では,最近の筆者の共同研究の成果から1)ショウジョウバエの産生する抗菌タンパク質に応答する,大腸菌側の遺伝子発現変化の解析7),2)シェーグレン症候群とIgG4関連疾患という症状がよく似た炎症性疾患の間での発現が異なる遺伝子の同定8)の二つの事例を,DNAマイクロアレイ解析の部分を中心に紹介する.
1)ショウジョウバエのペプチドグリカン受容体タンパク質による大腸菌遺伝子の発現誘導7)
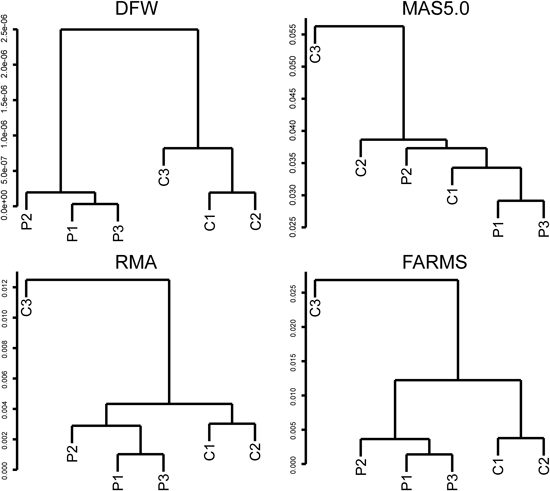
Kongらは,宿主に侵入した細菌は,宿主の免疫系からの作用によって自身の遺伝子発現を変動させる,という仮説を証明するために,ショウジョウバエの大腸菌感染モデルを用いた解析を行った.まず,ショウジョウバエのペプチドグリカン受容体-LCの細胞外領域および抗菌タンパク質attacinのGST(グルタチオンS-トランスフェラーゼ)融合リコンビナントタンパク質の混合物を大腸菌に添加し,DNAマイクロアレイを用いて大腸菌側の遺伝子発現変動を調べた.筆者はDNAマイクロアレイ実験およびデータ解析を担当し,上述のガイドラインを適用して発現変動遺伝子の抽出を行った.DNAマイクロアレイはAffymetrix社のGeneChip E. Coli Genome 2.0 Arrayを用いた.筆者は原核生物のGeneChipを扱うのは初めてであったため,これまでの解析手法がそのまま適用できるか,正直なところ若干の不安があったが,とにかくルーティンの解析を行った.経験的に成績のよいDFWに加え,MAS5.0, RMA, FARMS正規化を行った後,階層的クラスタリングを行った.その結果,DFW正規化データではショウジョウバエタンパク質添加群(P1, P2, P3)とコントロールのGST添加群(C1, C2, C3)がそれぞれクラスタを形成し,両者がきれいに分離した(図2).筆者がマイクロアレイ解析を行う際,最もほっとする瞬間である.このことから,これまで採用してきた正規化手法が原核生物由来のデータにも適用可能であることがわかった.その上で,ショウジョウバエタンパク質の添加によって大腸菌側に遺伝子発現変動が誘導されるという本論文における重要な結果をも示すことができ,さらには各群内の再現性もよいということが示されたわけである.図2に示したとおり,正規化手法によって結果が異なることがわかる.今回のデータからは,従来よく使われてきたMAS5.0のクラスタの分離は悪く,この先の解析に適さないこと,C3サンプルは全正規化手法に共通してやや離れた発現プロファイルを示すものの,DFWではGST添加群のクラスタ内に収まっている分,RMAやFARMSよりもこの先の解析を行うのに適していそうであること,等がみてとれる.なお,発現変動遺伝子の抽出には,DFWと相性がよいと上述のガイドラインで定められている,Rank products法を用いた.抽出した発現変動遺伝子セットに対して,GOアノテーションに基づくエンリッチメント解析を行った結果,代謝およびストレス応答に関する機能を示すGO termが濃縮されており,ショウジョウバエタンパク質添加によって大腸菌側に生理的変化が起こっていたことが考えられた.また,ショウジョウバエタンパク質の添加によって発現が上昇した133遺伝子のうち,宿主内での大腸菌の生存に直接関与しそうな31遺伝子を選んでRT-PCRを行った結果,17遺伝子の発現上昇が確認された.中でも,リポタンパク質をコードするnlpIは,変異体を用いたin vivo解析から,大腸菌がショウジョウバエに感染する際ペプチドグリカン受容体-LC依存的に発現上昇し,宿主中での大腸菌の生存期間延長に寄与していることが明らかとなった.
2)シェーグレン症候群とIgG4関連疾患の両者で発現の異なる遺伝子の同定8)
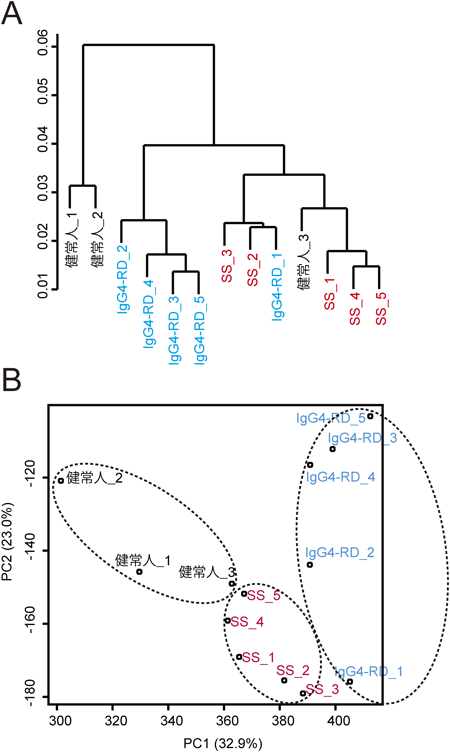
シェーグレン症候群(SS)は,自己免疫疾患の一種で,涙腺や唾液腺が自己免疫応答によって破壊されることによってドライアイ,ドライマウスといった症状が現れる.一方,IgG4関連疾患(IgG4-RD)も同様に涙腺や唾液腺に症状が出るが,SSとは異なる疾患である.診断基準が確立されたのも比較的最近のことであり18),現在も病因解明の途上にある難病の一つである.Tsuboiらは,IgG4-RDの病変局所における遺伝子発現を網羅的に解析することにより,IgG4-RDの病因・病態形成に関わる分子を見つけることを目的としてDNAマイクロアレイ解析を行った.筆者は臨床サンプルからのRNA調製を含むDNAマイクロアレイ実験およびデータ解析を担当した.DNAマイクロアレイはAffymetrix社のGeneChip Human Genome U133 Plus 2.0 Arrayを用いた.IgG4-RDは症例数がSSに比べると少ないため,3人分の口唇唾液腺検体を集めるのに2年を要した(リバイスの過程でさらに2検体追加され,最終的には各群5検体になっている).筆者はヒト臨床サンプルを扱うのはこのときが初めてで,しかも希少な検体のため重圧はあったが,結果的には問題なくデータをとることができた.この「問題なく」というのは,いくつかの基準をクリアしている必要がある.すなわち,抽出した全RNAの分解によるintegrityの低下がほとんどなく,マイクロアレイ用のサンプル調製に十分な量と濃度の全RNAが確保できていること,そして全RNAから調製したマイクロアレイ用サンプルのクオリティが十分でばらつきがないこと,である.今回の場合,DNAマイクロアレイ実験に進むことが可能な品質および量の全RNAが調製できたことから,臨床現場でのサンプリングが適切に行われていて,その後の筆者による抽出操作も問題がなかったことが証明された.さらには,上述したように正規化データのサンプル間クラスタリングの結果,ある程度仮説を支持するものである(この場合は「疾患ごとにクラスタが形成される」=「疾患間で遺伝子発現プロファイルが異なる」)ことも重要で,どのような正規化手法を試してみてもクラスタが分離しない場合は両群の遺伝子発現プロファイルは類似している,と結論せざるをえない.こちらに関しても,ルーティンの正規化,サンプル間クラスタリングを行い,確認した.階層的クラスタリングの結果だけみると,それぞれがクラスタを形成する傾向はあるものの,SSのクラスタに健常人・IgG4-RDそれぞれ1検体が入り込み,クラスタが明確に分離しているとはいえなかった(図3A).しかし,同じ正規化データを用いて主成分分析を行ってみると,群ごとにクラスタが分離し,両疾患の遺伝子発現プロファイルが異なっていることが明らかとなった(図3B).この段階で,DNAマイクロアレイ実験はほぼ成功したと判断できた.なお,階層的クラスタリングでSSのクラスタに含まれていた健常人_3, IgG4-RD_1の2検体は,主成分分析でみてもSSのクラスタに近いことがわかる.階層的クラスタリングも主成分分析も,やっていることは本質的には変わらないが,視点を少し変えると結果が異なる場合があるということである.
さて,こういった炎症性疾患の患部組織では,ベースとなる組織のみならず浸潤してきたマクロファージなどの細胞も含めての遺伝子発現をみることになるので,細胞のポピュレーションの違いをみているだけ,という可能性もあるが,それでもよいのである.つまり,「そのときにその組織で起こっていること」を,細胞のポピュレーションの変化をも含めてトランスクリプトームの変化として捉えているわけである.「遺伝子発現プロファイルが異なったのは,どのような細胞がどのように浸潤した結果なのか?」という疑問が生じれば,DNAマイクロアレイ解析の結果をもとにin situハイブリダイゼーションや免疫染色などで組織学的に確認すればよい.本研究では,「IgG4-RDの病因・病態形成に関わる分子を見つけること」を目的としていたため,DNAマイクロアレイで選抜された発現変動遺伝子について,さらに検体数を増やしてリアルタイムPCRで確認し,鍵となる分子を推定した.その結果,ケモカインCCL18をコードする遺伝子がIgG4-RDで高発現しており,病変局所に浸潤したマクロファージが産生するCCL18がT細胞・B細胞のケモタキシスを誘導していることが考えられた.
以上のように,バイオインフォマティクスの分野で提案された手法を積極的に取り入れた結果,DNAマイクロアレイだけでもかなりものがいえるようになったと感じる.もちろん,さまざまな角度からの検証は必要ではあるが,精度よい解析手法を選択することによって検証実験の効率化も図ることができている.網羅的解析の醍醐味は,先入観を排除することによって得られる新しい発見である.現在,生命科学研究に携わる研究者のほとんどが何らかの形でゲノム情報を利用していると思われるが,そこからもう一歩踏み込んで,網羅的解析から得られる大量のデータ処理に関してもぜひご自身でトライしていただきたい.
謝辞Acknowledgments
本稿を執筆するにあたり,ご推薦いただいた岩手医科大学いわて東北メディカル・メガバンク機構・清水厚志先生,ライフサイエンス統合データベースセンター・坊農秀雅先生に感謝申し上げます.また,本稿で取り上げたDNAマイクロアレイ解析手法の多くは,元同僚でもある東京大学大学院農学生命科学研究科アグリバイオインフォマティクス教育研究ユニット・門田幸二先生の作成されたウェブサイト「(Rで)マイクロアレイデータ解析」に依るところがきわめて大きく,また要所での彼の助言なくしては私の研究は成立しえませんでした.ここに心より感謝申し上げます.
引用文献References
1) Schena, M., Shalon, D., Davis, R.W., & Brown, P.O. (1995) Science, 270, 467–470.
2) Hubbell, E., Liu, W.M., & Mei, R. (2002) Bioinformatics, 18, 1585–1592.
3) Irizarry, R.A., Hobbs, B., Collin, F., Beazer-Barclay, Y.D., Antonellis, K.J., Scherf, U., & Speed, T.P. (2003) Biostatistics, 4, 249–264.
4) Hochreiter, S., Clevert, D.A., & Obermayer, K. (2006) Bioinformatics, 22, 943–949.
5) Chen, Z., McGee, M., Liu, Q., & Scheuermann, R.H. (2007) Bioinformatics, 23, 321–327.
6) Kohl, M. & Deigner, H.P. (2010) BMC Bioinformatics, 11, 583.
7) Kong, Q., Nakai, Y., Kuroda, N., Shiratsuchi, A., Nagaosa, K., & Nakanishi, Y. (2015) J. Biochem., 157, 507–517.
8) Tsuboi, H., Nakai, Y., Iizuka, M., Asashima, H., Hagiya, C., Tsuzuki, S., Hirota, T., Miki, H., Hagiwara, S., Kondo, Y., Tanaka, A., Moriyama, M., Matsumoto, I., Nakamura, S., Yoshihara, T., Abe, K., & Sumida, T. (2014) Arthritis Rheum. (Munch), 66, 2892–2899.
9) Suyama, T., Okada, S., Ishijima, T., Iida, K., Abe, K., & Nakai, Y. (2012) PLoS ONE, 7, e29483.
10) Kadota, K., Nakai, Y., & Shimizu, K. (2008) Algorithms Mol. Biol., 3, 8.
11) Breitling, R., Armengaud, P., Amtmann, A., & Herzyk, P. (2004) FEBS Lett., 573, 83–92.
12) Kadota, K., Nakai, Y., & Shimizu, K. (2009) Algorithms Mol. Biol., 4, 7.
13) R Development Core Team. (2008) R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria.
14) Gentleman, R.C., Carey, V.J., Bates, D.M., Bolstad, B., Dettling, M., Dudoit, S., Ellis, B., Gautier, L., Ge, Y., Gentry, J., Hornik, K., Hothorn, T., Huber, W., Iacus, S., Irizarry, R., Leisch, F., Li, C., Maechler, M., Rossini, A.J., Sawitzki, G., Smith, C., Smyth, G., Tierney, L., Yang, J.Y., & Zhang, J. (2004) Genome Biol., 5, R80.
15) Huang da, W., Sherman, B.T., & Lempicki, R.A. (2009) Nat. Protoc., 4, 44–57.
16) Binns, D., Dimmer, E., Huntley, R., Barrell, D., O’Donovan, C., & Apweiler, R. (2009) Bioinformatics, 25, 3045–3046.
17) Essaghir, A., Toffalini, F., Knoops, L., Kallin, A., van Helden, J., & Demoulin, J.B. (2010) Nucleic Acids Res., 38, e120.
18) Umehara, H., Okazaki, K., Masaki, Y., Kawano, M., Yamamoto, M., Saeki, T., Matsui, S., Sumida, T., Mimori, T., Tanaka, Y., Tsubota, K., Yoshino, T., Kawa, S., Suzuki, R., Takegami, T., Tomosugi, N., Kurose, N., Ishigaki, Y., Azumi, A., Kojima, M., Nakamura, S., & Inoue, D. (2012) Mod. Rheumatol., 22, 1–14.
著者紹介Author Profile
 中井 雄治(なかい ゆうじ)
中井 雄治(なかい ゆうじ)弘前大学食料科学研究所教授.博士(農学).
略歴1989年東京大学農学部農芸化学科卒業.96年同大学院農学生命科学研究科応用生命化学専攻博士課程修了.国立衛生試験所,理化学研究所ポスドクを経て2000年金沢大学薬学部助手.05年東京大学大学院農学生命科学研究科アグリバイオインフォマティクス人材養成ユニット特任助教授,09年同大学院同研究科ILSI Japan寄付講座「機能性食品ゲノミクス」特任准教授を経て14年より現職.
研究テーマと抱負摂取した食物に対する生体応答の網羅的解析を通じて,生体の恒常性調節の仕組みを解明したい.また,ニュートリゲノミクスの手法を用いて様々な農林水産物から新たな生理機能を見つけたい.
ウェブサイトhttp://www.ifs.hirosaki-u.ac.jp
趣味ジャズ(トロンボーン演奏),自転車,ウォーキング.