東北大学東北メディカル・メガバンク機構(以下,ToMMo)は,岩手医科大学とともに東日本大震災からの復興事業として,東北メディカル・メガバンク計画に取り組んでおり,宮城県および岩手県の地域住民15万人規模のゲノムコホート調査を2013年から実施している.筆者らの所属するゲノム解析部門では高性能シークエンサー(high throughput sequencer,以下,HTS)を活用してコホート参加者の全ゲノム配列の解析を行い,日本人の詳細な遺伝的多様性を明らかにした1).HTSはがんや遺伝性疾患などのゲノム医学研究に活用されることが多いが,今回我々が行ったのは,同調査に参加同意いただいた日常生活を一般に過ごすことができる「健常人」の全ゲノム配列解析である.まずは健常人の日本人の全ゲノム解析を大規模に行う目的と意義を述べたい.
ヒトの疾患は,各個人が持つ遺伝的背景の影響を受けるものが多い.たとえば2型糖尿病の20%以上は遺伝的背景で説明できると見積もられている.とりわけ,生活習慣病など比較的罹患率の高い疾患は複数の遺伝的変異が発症のリスクに関連しており,個々の遺伝的変異の多くは疾患を持つ人と持たない人の間で共有されていることが多い.このような疾患の原因遺伝子の探索にはゲノムワイド関連解析(genome-wide association study:GWAS)が有効であり,ここ数年で数多くの疾患関連遺伝子の同定が報告されてきた.一方で全ゲノム規模の解析にも関わらず疾患原因の遺伝的寄与すべてを解明するには至っておらず,GWASで同定できた疾患と関連する頻度が高い一塩基多型(single nucleotide polymorphism,以下,SNP)情報(本稿では,変異は集団中ですべての頻度でみられる多様性を指し,多型の場合には特に集団中で頻度1%以上の変異を意味する)だけでは多因子疾患の遺伝率は十分に説明できないことが明らかになってきた(“失われた遺伝率”).HTSを使った全ゲノム解析は,従来の全ゲノム中の一部のSNP情報を取得するSNPアレイでは発見不可能な挿入欠失やコピー数変異などの構造変異や集団中で1%以下の低頻度の変異までも解析可能であり,失われた遺伝率を埋める切り札の一つになると考えられている.しかし,HTSの問題点には解析にかかるコストがあげられる.近年HTSを用いた解析は劇的に安価になったが,依然,解析に必要な計算機資源のコストなどを含めると1検体あたり20~30万円が必要であり,SNPアレイの解析コストとは10倍近い開きがある.また大規模な全ゲノム解析には検体管理,シークエンス,データ解析用のスーパーコンピュータなど大規模な設備の構築が不可欠であり,解析に多数の検体を要する多因子疾患の責任変異の探索にHTSを使った全ゲノム配列解析を行うことは現時点では現実的ではない.
そのため,多因子疾患の研究では対象とする遺伝集団の健常人の遺伝的多様性をあらかじめカタログ化した「リファレンスパネル」を個別の研究で構築し,参照するというアプローチがとられている.「国際1000人ゲノム計画」では,当時普及し始めていたHTSを大規模に活用し,全人類が持つ比較的頻度の高いSNPや構造多型を網羅的に発見するとともに,全ゲノム規模のリファレンスパネルを最初に構築した.国際1000人ゲノム計画の全ゲノムリファレンスパネル(以下,1KGPパネル)2)は14地域の人類集団,合計1092人の全ゲノム配列から構築されている.しかし,1KGPパネルは世界各地の集団から構成されるため,含まれる個々の集団は多くても150人程度のサンプルしか含まれていない.たとえば,1KGPの日本人サンプルの場合,JPT(Japanese in Tokyo)の89人分しか含まれていない.そこで我々は,HTSを用いた1070人の高精度な全ゲノム解析を行い,日本人が持つ構造変異やマイナーアレル頻度(以下,MAF)0.5%までの変異を網羅した日本人全ゲノムリファレンスパネルの構築を行った.
同パネルを後述する遺伝子型インピュテーション法を用いることでゲノムコホートでの数十万人規模のSNPアレイによるタイピングと組み合わせることが可能となり,数十万人規模のコホート参加者の頻度1%までのSNP情報をほぼ網羅した全ゲノム情報を低コストで効率よく取得することができる.同様の手法はイギリスのUKバイオバンクなどでも行われており,生活習慣やその他,環境要因とゲノム情報交互作用による病気の発症リスクの予測など個別化予防,医療に埋めた有効な基盤情報になっていくと考えている.
本稿では1KJPNパネルの構築,得られた変異情報の詳細や情報解析結果,さらに情報解析のために開発したバイオインフォマティクスツールについて報告をする.なお,さらなる詳細については筆者らの原著論文を参照されたい1).
東北メディカル・メガバンク機構のコホート参加者から1344人の解析候補を抽出し1KJPNパネルの構築を進めた.選択にあたっては,追跡可能であり,DNA量がSNPアレイおよびHTSのために十分にあり,かつDNA品質が一定基準をクリアした検体を選択した.すべての検体は同意取得がなされており,解析にあたっては匿名化の後に行った.すべてのDNAサンプルはIllumina社のHumanOmni2.5で解析を行い,近親者や一定基準を満たさなかったサンプルを除外することで最終的に1201人のHTS解析を行った.HTSにあたり,HiSeq2500のPCRを前処理で行わないプロトコル(以下,PCR-freeプロトコル)を用いて約550塩基に断片化された染色体の各断片1両端162塩基ずつを読み,各サンプルは平均32.4倍の高深度で情報取得を行った.最終的に,1201人のうち1070人を全ゲノムリファレンスパネルの検体として採用し,残り131人については同パネルの検証用として利用することとした.なお,1070人から,合計で100.4兆塩基ものシークエンスを取得した.さらに,読み取った後のシークエンスは筆者らが開発を行ったSUGARという品質チェック用のソフトウェアで精度確認を行った.
リファレンス配列には国際参照配列のGRCh37/hg19にデコイ配列(hs37d5)を組み込んだ参照配列を利用し,同参照配列へのアライメント(シークエンシングの一つ一つの読み取り結果が参照配列のどこ由来かを推定すること)と変異コール(各個人の変異の箇所とパターンを推定すること)は,複数のソフトウェアを組み合わせて解析を行った.その結果,2960万個のSNV(high-sensitive SNVs set),約197万の短い欠失変異(72.6%が新規),約138万の長い挿入変異(75%が新規),47,343個の100塩基以上の欠失変異,9354個の100塩基以上の挿入変異を常染色体上に見つけることができた.さらに,信頼度の高いSNVを取得するために,ソフトウェア解析依存で発生する傾向,ハーディー・ワインベルグの法則を逸脱するSNV,深度のばらつきが多い領域などを考慮した複数のフィルタリング処理を行うことで,信頼度の高い合計2120万個のSNV(うち56.6%が新規の変異)を構築した(high-confidence SNVs set,表1,以下,断りがない場合,このSNVの集合を1KJPNパネルと呼ぶ).この新規変異の割合は,オランダやアイスランドなど他のリファレンスパネルと同様の傾向であった.サンガーシークエンスやマスアレイを用いてfalse discovery rate(以下,FDR)の検証を行ったところ,SNVは174個検証して0個(FDR 0%;信頼区間0.0~1.10%),短い欠失は32個検証して0個(FDR 0%;信頼区間0.0~5.78%),短い挿入は22個検証して1個(FDR 3.85%;信頼区間0.49~19.34%)という好成績であった.また,カスタムアレイを作成して別の方法でSNVの精度検証をしたところFDR 0.8%;信頼区間0.63~0.97%という結果が得られた.なお,推定FDRはMAFに強い依存がないことも観測されており,1KJPNのSNVの精度は,アレル頻度にあまり依存しない結果であるといえる.
表1 1KJPNのSNV, INS, DELのまとめ | 総数 | | 1017 |
| 総塩基数 | | 100.4兆塩基 |
| 平均シークエンス深度 | | 32.4x |
| high-sensitive SNVs | high-confidence SNVs |
| SNVs | 総数 | 29,588,649 | 21,221,195 |
| 既知の数 | 12,308,520 | 9,219,783 |
| 新規の数 | 17,280,129 | 12,001,412 |
| 新規の割合 | 58.40% | 56.55% |
| サンプルごとの平均総数 | 3,886,081 | 2,716,853 |
| サンプルごとのヘテロ接合総数 | 2,252,841 | 1,532,773 |
| 長さ | 1 bp≤長さ<100 bp | 100 bp≤長さ |
| 欠失 | 総数 | 1,969,302 | 47,343 |
| 新規の数 | 1,429,636 | — |
| 新規の割合 | 72.60% | — |
| インフレーム/フレームシフトの数 | 3112/4454 | — |
| サンプルごとの平均総数 | 190857 | 2654 |
| 長さ | 1 bp≤長さ<100 bp | 100 bp≤長さ |
| 挿入 | 総数 | 1,384,230 | 9354 |
| 新規の数 | 1,037,839 | 9354 |
| 新規の割合 | 74.98% | — |
| インフレーム/フレームシフトの数 | 1577/2506 | — |
| サンプルごとの平均総数 | 159,359 | 45 |
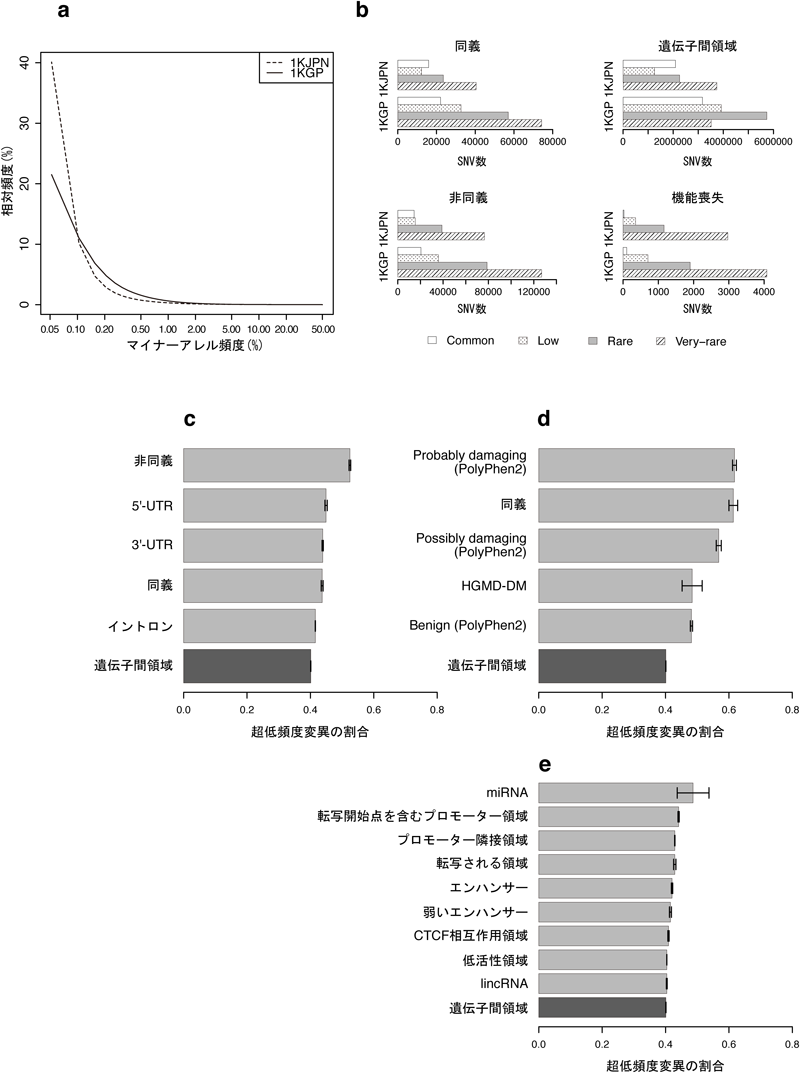
ゲノムDNAのPCR増幅やターゲットキャプチャによるエクソーム領域に限定した解析はシークエンシングの過程によって固有な偏りやエラーを生じうる原因になりうる.本研究では前述のとおり,PCR-freeプロトコルを用いた全ゲノムシークエンシングを行うことで,これらに起因するSNVの誤検出を可能な限り抑えることを目指した.図1aは1KGPパネルと1KJPNパネルの相対的なアレル頻度を比較しているが,1KGPパネルは低頻度変異の相対的な頻度が1KJPNパネルに比べて明らかに低いのがわかる.これは遺伝子間領域など1KGPにおいて低深度のシークエンシングしか行われていない領域において低頻度変異を補足できていないことを反映している.実際,1KGPパネルの総SNV数は1KJPNパネルに比べて多いにも関わらず,遺伝子間領域のSNV数は1KJPNパネルの方が多い(図1b).
領域間に偏りのない精密なアレル頻度分布は領域間の有害変異の蓄積度合いの違いをみるよい指標になる.ここで有害変異とは数世代~数十世代にわたって集団内の遺伝子頻度に効果を及ぼすようなものを指す.ヒト集団に生じた新規突然変異は一定時間集団内に存在するが,有害度の高い変異ほど集団から早く取り除かれる.その結果として,有害な突然変異ほど集団中で低頻度に存在することになる.集団中のアレル頻度は集団サイズなどの他の要因によっても影響を受けうるが,本研究のように同一集団に属するサンプルの場合このような要因はゲノム全体に及ぶので,領域間の相対的なアレル頻度分布の違いは各領域の機能的な差異を反映しているとみなせる.この考えのもと領域間の機能的な差異,つまり負の自然淘汰の影響の評価を行った.そこで,ある機能カテゴリで発見されたSNVのうち1KJPNパネル内でMAFが0.5%未満のSNVの割合をFVRV(fraction of very-rare variant,超低頻度変異の割合)と定義して領域間の比較を行った.FVRVの値が低いほどその機能カテゴリ内のSNVは負の自然淘汰を受けている,つまり有害な変異が集積しているということになる.図1c, dはさまざまな分類基準で定義した機能カテゴリごとのFVRVを比較したものである.図1cは遺伝子を構成するUTR,イントロン,エクソンに加え遺伝子間領域のFVRVを示しており,エクソン領域はアミノ酸置換を伴う非同義SNV(nonsynonymous SNV)と伴わない同義SNV(synonymous SNV)に分けた.遺伝子間領域のFVRVが最も低く(0.40),これはこの領域の大部分が機能を持たず中立的な変異が集団中に蓄積していることを支持している.アミノ酸置換を伴う非同義変異はタンパク質機能の変化を伴うことが多く,その大部分は有害であることが過去の研究でも示されてきた3)が,本研究でも高いFVRVが観察された.興味深いことに他の遺伝子領域(5′-UTR, 3′-UTR,同義SNV)も遺伝子間領域よりも高いFVRVが観察され,弱い負の自然淘汰の影響下にあることを示している.同義SNVはアミノ酸置換を伴わないもののタンパク質の翻訳効率に影響しそれが弱い負の自然淘汰を引き起こしているとの研究4, 5)があるが,この結果はこの仮説を支持すると考えられる.
図1dは非同義置換の結果生じるアミノ酸置換の効果予測(PolyPhen2とSIFT),機能喪失型変異(loss of function mutation),疾患を引き起こすことが報告されている変異(HGMDデータベースによる)のFVRVを比較したものである.PolyPhen2で強い効果が予測される変異ほどFVRVも高い値を示しており,最も効果の強いカテゴリ(probably damaging)のFVRVは機能喪失型変異とほぼ同じFVRVであった.一方,HGMDデータベースで「疾患を引き起こす突然変異」カテゴリ(DM:disease mutation)のFVRVはPolyPhen2の予測で最も効果の弱いBenignと同等のFVRVであった.
図1eは非タンパク質遺伝子領域のFVRVを比較した結果である.ノンコーディングRNA(miRNAとlincRNA)とENCODEプロジェクトの結果に基づき予測された機能性エレメントでカテゴリ分けを行った.図1eの中でmiRNAはFVRVが最も高く,この領域の変異は遺伝子領域に匹敵するインパクトを持つことが示唆される.また,ENCODEで活性がほとんどないと予測される低活性領域(repressed or low activity region)のFVRVが最も低く,それに比べ転写活性やプロモーター領域は有意に高いFVRVがあることが示された.
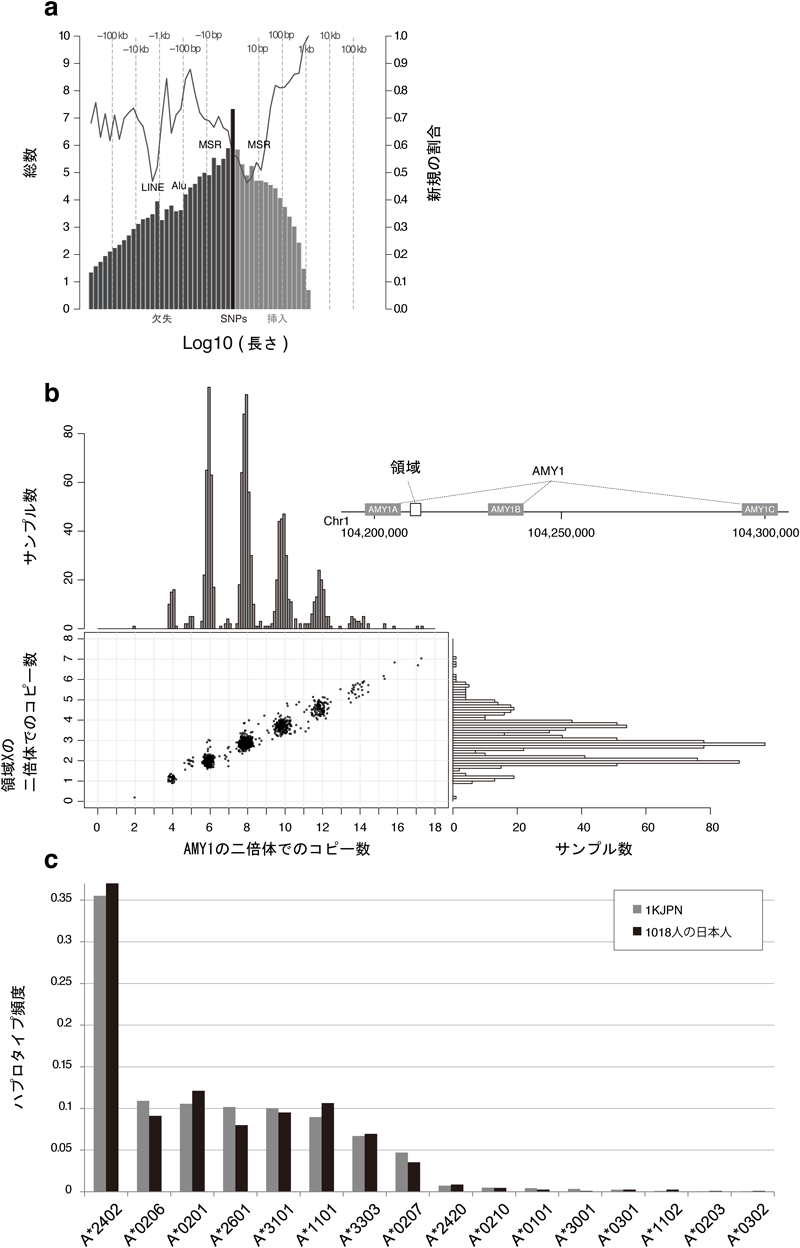
高深度のHTSデータを用いることによって,各個人が持つ挿入・欠失変異(以下,INS・DEL)やコピー数変異(以下,CNV)を網羅的にタイピングすることができた.図2aでは1KJPNの1070人における区別できるINSおよびDEL箇所の個数を長さごとに示しており,サイズの大きい変異の方がより頻度が低いという傾向が現れている.またLINE/Alu配列にも頻度のピークがみられ,これらの傾向は1KGP2)やオランダ・ゲノム6)の結果と整合している.検出された50塩基以上の長いINSのほとんどは新規であり,高深度のHTSデータによる検出の有効性が示されている.100塩基以上のINSは同じ長さのDELと比較して検出数が少ないが,今後長鎖型のシークエンサーの活用によって明らかになると考えられる.
CNVの検出においては,HTSデータのアライメントされるリード数がコピー数にほぼ比例する関係を利用する.高深度データはコピー数の高い分解能を与えるため,CNVの解析にも有用である.今回の解析によって1KJPN集団における25,923のCNV箇所を特定することができた.その中で,特にデンプンの消化に関わるアミラーゼ遺伝子(AMY1)の平均コピー数は8.27であり,デンプン消費量が少ない集団における平均値5.44よりも顕著に高いことが明らかになった(図2b).この結果は,デンプン消費量の多い集団において,AMY1のコピー数が増加しているという過去の研究結果7)と整合するものである.さらに興味深いことに,AMY1の二倍体コピー数が集団の大多数で奇数になっていることから,AMY1の一倍体コピー数の増加単位が2であろうことが予想された.我々は実際にリファレンスゲノム上のAMY1AとAMY1Bの間にある領域の二倍体コピー数nがAMY1の二倍体コピー数yとy=2n+2の関係にあることを確認し,過去に提唱されたAMY1AからAMY1Bの領域がコピー数の増加単位になっているという仮説8)を裏づけることができた.遺伝子領域におけるCNVのコピー数は遺伝子の発現量とほぼ正の相関があることがわかっており,今後の表現型や疾患との関連解析などに活用が可能である.
高深度HTSデータは変異の多様性が大きいアレルの同定にも有用である.ヒト白血球抗原(human leukocyte antigen:HLA)は多様性が大きく,ハプロタイプ構造がヒト集団間で異なっていることが知られている9).本研究においては,開発したHLA-VBSeq10)を用いて1KJPNのHLA-A,-B,-Cのアレル頻度を同定した.HLA-VBSeqはIMGT/HLAデータベースに登録されたHLA領域にアライメントすることでタイピングを行っている.HLA-Aについては1KPJNのほとんどのアレル(2140のうち2063)を最大解像度である8-digitで決定できた.また,1KJPNにおけるHLA-A,-B,-Cの4-digitまでの頻度は,PCR-SSOPを用いて1018人の日本人からタイピングした既知の頻度11)と非常に近いものであった(図2c).HLA遺伝子型が重要となる領域は,臓器移植や感染症への感受性,自己免疫疾患など多岐にわたる.本研究で示したような正確なタイピングを行うことは関連解析や医療現場における患者とドナーのマッチングにも重要である.
1)フェージング
シングルトン変異(1KJPNパネル中に一つしかない変異のこと)を除いた1KJPNの遺伝子型情報を,SHAPEIT212)によりフェージング(2本の染色体上の連続したアレルの並びを推定)することで,フェーズ済みリファレンスパネルの作成を試験的に行った.SHAPEIT2では,各ハプロタイプは他のハプロタイプにおける変異ならびに組換えにより構成されることを仮定したPAC尤度と呼ばれる原理を元にフェージングが行われるが,頻度の低い変異についてはフェージングの推定精度が低く,特にシングルトン変異については,各個人について二つのハプロタイプのうちのどちらに属するかがランダムに推定されるため,ここではフェージング対象から除いている.1KJPNの遺伝子型情報としては,high-sensitive SNVs setに対して短いゲノム挿入変異および欠失変異を加えた変異データセットを用いた.SHAPEIT2のフェージングデータに対して,下記の三つの手順によりシングルトンについてフェージングを行った.
- ①複数箇所の変異をカバーするリードから局所的なフェージングを行う.HapMonster13)により,局所的なハプロタイプ情報を得ることができ,シングルトン変異についてもフェージング情報を得た.
- ②局所的なハプロタイプ情報がSHAPEIT2から得られたハプロタイプとフェーズ情報について矛盾がない場合,局所的なハプロタイプ情報を元に,シングルトン変異がフェージングした形で,フェーズ済みリファレンスパネルに組み込まれる.一方,局所的なハプロタイプ情報とSHAPEIT2からの結果が矛盾する際には,該当の領域に含まれるシングルトン変異の情報はフェーズ済みリファレンスパネルには組み込まれない.
- ③局所的なハプロタイプ領域に含まれないシングルトン変異についても,フェーズ済みリファレンスパネルには含めない.
上記の結果,43%のシングルトン変異がフェーズ済みリファレンスパネルに組み込まれた.
2)遺伝子型インピュテーション
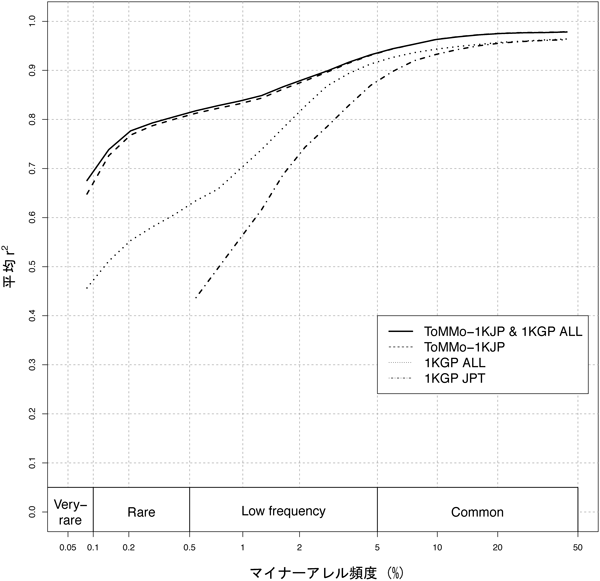
フェーズ済みリファレンスパネルの利活用法の一つとして,計測された遺伝子型から周囲の未計測の遺伝子型の推定を行う遺伝子型インピュテーション(以下,インピュテーション.ある個人の数十万のSNPタイピングの結果に対して,リファレンスパネルを用いることで,その個人がもつその他のSNPを全ゲノム領域にわたって計算機上で推定すること)がある.ここでは,1KJPNパネルのサンプルとは独立な131人の日本人サンプル(詳細は2節参照)について,SNPアレイであるHumanOmni2.5-8 BeadChipにおいて設計されている箇所の遺伝子型情報に対してIMPUTE2(ver.2.2.2)14)を用いてインピュテーションを行い,その推定精度を解析した.IMPUTE2では,フェーズ済みリファレンスパネルを元にインピュテーションが行われるが,1KJPNに加え,国際1000人ゲノムプロジェクトより2013年12月にリリースされた1092人の多民族からなるフェーズ済みリファレンスパネル(以下,1KGP),また,1KGPの部分リファレンスパネルである89人のHapMap JPTサンプルからなるフェーズ済みリファレンスパネル(以下,1KGP-JPT)の三つのフェーズ済みリファレンスパネルを用いて性能評価を行った.評価方法としては,正解とされる遺伝子型と推定された遺伝子型を数値化し,その決定係数(r2)を評価値とする文献14)記載の方法を用いた.具体的には,各変異サイトにおける二つの対立遺伝子型A, aについて,AA, Aa, aaの3種の遺伝子型が考えられるが,それぞれに対して0, 1, 2の3値の割り当てを行った.また,インピュテーション推定結果として,3種の遺伝子型への事後確率が得られるが,この事後確率により期待値をとった値であるallele dosageと正解の遺伝子型に対応した値の間で決定係数を計算した.ここでは,シークエンシングデータから同定された遺伝子型を正解として用いた.図3は,MAFを一定の範囲で区切り,1KJPNと1KGPの双方において含まれているSNVを対象として計算されたr2値について各ビンにおいて平均化された値のプロットである.図のプロットにおけるMAFは,各リファレンスパネルにおいて独立に計算された値である.1KJPNにおけるr2値は,MAF全域において他のリファレンスパネルと比べ高い値となっており,特にMAFの低い領域においては,その性能差は大きく,日本人サンプルへのインピュテーションにおける,1KJPNを元にしたフェーズ済みリファレンスパネルの有用性が確認できた.
1KJPNの変異について,遺伝子情報,変異の効果予測,そして既知の疾患変異情報のアノテーションを通して,遺伝子機能への効果が大きいものや,疾患との関係が知られている変異を同定できた.既知の疾患変異情報のリソースとして,The Human Gene Mutation Database(HGMD)を利用した.一人あたり,疾患原因変異(HGMDが定義しているdisease-causing mutation)を平均で11.2個(ヘテロ接合で9.6個,ホモ接合で1.6個)持っていることがわかった.同様に,途中で終止コドンが生じるナンセンス変異の存在量を計算したところ,一人あたり平均で50~65個持っていることがわかった.これらの推定量は,他の東アジア集団での推定量とほぼ同等であった.ナンセンス変異は全体で3505個同定されているが,そのうち既知の疾患原因変異に相当するものの割合は4.5%であった.これは,ナンセンス変異の大多数は,医学・生物学的効果がまだ不明であることを示唆する.
遺伝病の発見率は集団ごとに異なっており,たとえば日本人での先天性代謝異常の発見率は,欧米よりも低いことが知られている.集団ごとのリスクアレルの頻度情報は,現実の罹患率のデータと比較することによって,浸透率の推定や,他の要因があるかどうかの検討に有用である.HGMDの変異に相当するSNVについて頻度を集団間(国際1000人ゲノムの14集団)で比較したところ,2638の変異について,集団間の有意な頻度差がみられた.特に,FUT2遺伝子のSNP rs1047781は,遺伝子産物(FUT2酵素)の129番目のアミノ酸にイソロイシンあるいはフェニルアラニンの違いを起こすのである.フェニルアラニン型のアレル頻度は,1KJPNでは0.38であったが,ヨーロッパ集団では0であった.FUT2酵素はABH抗原を唾液中にも分泌させる酵素であり,古典的なsecretor locusとして知られている.この変異のホモ接合体の頻度は0.141であり,これは「約15%の日本人が,非分泌型である」という報告とほぼ合致する.最近の研究では,このSNP rs1047781は,腫瘍バイオマーカーとの関連が報告されている.
8. リファレンスパネルの作成にあたり開発したバイオインフォマティクスツール群
現在,HTSデータ解析に使われているツールの多くは大規模シークエンスプロジェクトの過程で開発され公開されたものである.本プロジェクトにおいてもさまざまなツールの開発を行いリファレンスパネルの構築に役立て公開している.最後にここでこれらのツールを簡単に紹介させていただく.詳細については,各原著論文にあたることをお勧めする.
1)SUGAR
SUGAR15)はHiSeqなどのシークエンスの品質を可視化するとともに物理的なフローセルの位置情報やアライメントの悪いタイルなどさまざまな条件でシークエンス情報をフィルタリングすることができるJavaで実装されたツールである.我々はリファレンスパネル構築の品質チェックのために開発し利用した.可視化ツールとしてHTQCやSolexaQAがあるが,これらのツールでは扱えない気泡混入などの偶発的エラーのレベルについても可視化およびフィルタリングを行うことができる.このツールにより,微量ながん細胞が正常細胞に混入しているシークエンス結果など,できる限り高品質なシークエンスの抽出を行いたい場合などにも利用可能である.
2)HapMonster
HapMonster13)は,HTSデータから変異コールと局所的なフェージングを同時に行うソフトウェアである.局所的なフェージングについては,複数のヘテロ接合変異サイトをまたぐシークエンスリードを用いて推定を行う.Javaで実装されており,http://nagasakilab.csml.org/en/hapmonsterにてjar形式のソフトウェアのダウンロード可能である.
3)iSVP
iSVP16)は,HTSデータから構造変異(SV)を検出する複数のツールを並列に適用し,結果を統合するパイプラインである.挿入変異についてはGATK Haplotype Caller(HC)の結果を用い,欠失変異については,BreakDancer, Pindel, GATKとHCの予測精度を変異の大きさごとのシミュレーション評価に基づいて適切な統合を行う.リファレンスパネルの構築にはiSVPを利用している.
4)HLA-VBSeq
HLA-VBSeq10)は,HTSデータから変分ベイズ推定によってHLAの型を8桁の精度で同定することができるソフトウェアである.今後HLAのリファレンス配列情報が充実することでさらに精度向上が見込める.
5)iJGVD
integrative Japanese Genome Variation Database(iJGVD, http://ijgvd.megabank.tohoku.ac.jp/)17)では,ToMMoの1070人分の全ゲノム解読から得られた変異の頻度情報を公開している.2015年10月現在,アレル頻度5%以上の一塩基多型頻度情報約430万件について公開している(2015年12月末には,すべての頻度の一塩基多型頻度情報を公開予定).rsSNP IDや遺伝子シンボルで検索することや国際ゲノム参照配列上での位置情報の把握などができる.また,ジャポニカアレイ(日本人の持つSNVのうち約65万個を搭載したアレイ.インピュテーションとリファレンスパネルを用いることで全ゲノム領域の変異を高精度で推定できる.次世代型アレイの一つ)で設計されている変異についても検索することができる.これらのデータセットについては,NBDCヒトデータベースからも公開し,一括ダウンロードが可能である(データID:hum0015).
今回東北メディカル・メガバンク機構のゲノムコホート研究の参加に同意いただいた1070人の全ゲノム配列の解読を高深度で行い,全ゲノムリファレンスパネル(1KJPNパネル)の構築を行った.また,日本人を対象としたゲノム医学研究の研究基盤として広く利活用されるように,平成27年8月より後述のhigh-confidence SNVs setなどの情報分譲を開始している.
2120万個のうち約1200万個はこれまでに報告のなかった新規のSNVであるが,これは日本人集団が持つと期待される変異のどの程度をカバーしているのだろうか? 過去の日本人集団の人口変動を考慮した集団遺伝学モデルを用いるとMAF 0.1%以上のSNVのうち99%以上が今回の解析で発見されたと見積もられる.ただし,この結果は反復配列など十分なシークエンス精度が期待されない領域を除外した結果であるので,これらの領域を含めると実際の発見率はもう少し低いと考えられる.また,今回は東北地方のサンプルでの解析であるので,他の地域の集団に固有な変異もリファレンスパネルには含まれていない.今後は,リファレンスパネルのサンプルに他の地域の全ゲノム解析結果も追加することで,さらなる拡充を行うことを計画している.全ゲノムリファレンスパネルの拡充は日本人集団を対象としたジェノタイプインピュテーションのさらなる精度向上が期待されるばかりではなく,希少な遺伝性疾患の罹患率推定など遺伝医学研究に重要な寄与を行えると考えている.
全ゲノムシークエンスを高深度で行うことによってさまざまな構造変異(indel, CNV)を集団単位で検出することができた.ただし短鎖型のハイスループットシークエンサの技術的な制約で100塩基を超える挿入変異やセントロメアやテロメア配列など長い反復配列中の変異は十分に発見できていないものと思われる.このようなタイプの変異を発見するためには長鎖型のシークエンサーの活用が今後必要不可欠である.
HTSによる全ゲノム解析はそのコストも十分に下がり,遺伝医学研究の重要なツールになったといえる.また,今回の解析で示されたように十分な深度で解析を行えば希少疾患の原因変異候補も検出できる.その際にSNVの頻度を正確に与える全ゲノムリファレンスパネルは大いに役立つものと期待される.また,本研究では全ゲノムリファレンスパネルが高精度な全ゲノムジェノタイプインピュテーションを行う上でも有用であることを示した.生活習慣病などのありふれた疾患においてはMAF 5%以上のSNPに加えてMAF 0.5%から5%までの希少な変異も重要な寄与をしてきていることが近年明らかになってきている.このような変異の有意性をゲノムワイド関連解析で示すために近年では1万サンプル以上を用いた大規模解析が行われることも珍しくない.このような大規模解析においてはHTSの利用はコスト的に現実的ではなくSNPアレイを活用しなければならない.今回示したように全ゲノムリファレンスパネルを活用した高精度な全ゲノムインピュテーションは,HTSとSNPアレイのコスト的なギャップを埋める有効な手段である.そこで我々は1KJPNパネルを使った全ゲノムインピュテーションを効果的に行うために1KJPNパネルに基づき日本人向けSNPアレイ「ジャポニカアレイ®」を設計して活用を行っている1).
謝辞Acknowledgments
当研究は,東北メディカル・メガバンク事業(東日本大震災復興特別会計)として行われました.
本総説に記載している主な内容は文献1に記載しています.また,当総説の著者は総説の執筆者としましたが,本リファレンスパネルの構築は,東北大学東北メディカル・メガバンク機構における地域医療支援,コホート,バイオバンク,シークエンス解析,ICT情報管理などさまざまな研究者および支援者の貢献により初めて達成することができました.関係したメンバについては,http://www.megabank.tohoku.ac.jp/english/a141201/を参照ください.また文献1の共著者は以下となります.安田純先生,勝岡史城先生,成相直樹先生,横澤潤二先生,檀上稲穂先生,齋藤さかえ先生,佐藤行人先生,津田薫先生,齋藤るみ子先生,潘小青先生,西川慧先生,伊藤信先生,黒木陽子先生,田邊修先生,布施昇男先生,栗山進一先生,清元秀泰先生,寳澤篤先生,峯岸直子先生,James Douglas Engel先生,木下賢吾先生,呉繁夫先生,八重樫伸生先生,坪井明人先生,長神風二先生,川目裕先生,富田博秋先生,辻一郎先生,中谷純先生,菅原準一先生,鈴木吉也先生,菊谷昌浩先生,阿部倫明先生,中谷直樹先生,大隅典子先生,山下理宇先生,荻島創一先生,高井貴子先生,冨永悌二先生,瀧靖之先生,鈴木洋一先生,山本雅之先生.
すべての計算リソースはToMMoスーパーコンピュータシステムを使って行いました.(http://sc.megabank.tohoku.ac.jp).また,リファレンスパネルの構築について助言をいただいた岩手医科大の岩手メディカル・メガバンク機構のすべての方々,特に祖父江憲治先生,人見次郎先生,清水厚志先生に感謝します.最後に,東北メディカル・メガバンク機構のコホート調査に参加いただいたすべての参加者に深く感謝いたします.
引用文献References
1) Nagasaki, M., Yasuda, J., Katsuoka, F., Nariai, N., Kojima, K., Kawai, Y., Yamaguchi-Kabata, Y., Yokozawa, J., Danjoh, I., Saito, S., Sato, Y., Mimori, T., Tsuda, K., Saito, R., Pan, X., Nishikawa, S., Ito, S., Kuroki, Y., Tanabe, O., Fuse, N., Kuriyama, S., Kiyomoto, H., Hozawa, A., Minegishi, N., Douglas Engel, J., Kinoshita, K., Kure, S., Yaegashi, N., To, M.J.R.P.P., & Yamamoto, M. (2015) Nat. Commun., 6, 8018.
2) Genomes Project, C., Abecasis, G. R., Auton, A., Brooks, L. D., DePristo, M. A., Durbin, R. M., Handsaker, R. E., Kang, H. M., Marth, G. T., & McVean, G. A. (2012) Nature, 491, 56–65.
3) Fu, W., O’Connor, T.D., Jun, G., Kang, H.M., Abecasis, G., Leal, S.M., Gabriel, S., Rieder, M.J., Altshuler, D., Shendure, J., Nickerson, D.A., Bamshad, M.J., Project, N.E.S., & Akey, J.M. (2013) Nature, 493, 216–220.
4) Ikemura, T. (1985) Mol. Biol. Evol., 2, 13–34.
5) Powell, J.R. & Moriyama, E.N. (1997) Proc. Natl. Acad. Sci. USA, 94, 7784–7790.
6) Genome of the Netherlands, C. (2014) Nat. Genet., 46, 818–825.
7) Perry, G.H., Dominy, N.J., Claw, K.G., Lee, A.S., Fiegler, H., Redon, R., Werner, J., Villanea, F.A., Mountain, J.L., Misra, R., Carter, N.P., Lee, C., & Stone, A.C. (2007) Nat. Genet., 39, 1256–1260.
8) Groot, P.C., Bleeker, M.J., Pronk, J.C., Arwert, F., Mager, W.H., Planta, R.J., Eriksson, A.W., & Frants, R.R. (1989) Genomics, 5, 29–42.
9) Itoh, Y., Mizuki, N., Shimada, T., Azuma, F., Itakura, M., Kashiwase, K., Kikkawa, E., Kulski, J.K., Satake, M., & Inoko, H. (2005) Immunogenetics, 57, 717–729.
10) Nariai, N., Kojima, K., Saito, S., Mimori, T., Sato, Y., Kawai, Y., Yamaguchi-Kabata, Y., Yasuda, J., & Nagasaki, M. (2015) BMC Genomics, 16(Suppl 2), S7.
11) de Bakker, P.I., McVean, G., Sabeti, P.C., Miretti, M.M., Green, T., Marchini, J., Ke, X., Monsuur, A.J., Whittaker, P., Delgado, M., Morrison, J., Richardson, A., Walsh, E.C., Gao, X., Galver, L., Hart, J., Hafler, D.A., Pericak-Vance, M., Todd, J.A., Daly, M.J., Trowsdale, J., Wijmenga, C., Vyse, T.J., Beck, S., Murray, S.S., Carrington, M., Gregory, S., Deloukas, P., & Rioux, J.D. (2006) Nat. Genet., 38, 1166–1172.
12) Delaneau, O., Zagury, J.F., & Marchini, J. (2013) Nat. Methods, 10, 5–6.
13) Kojima, K., Nariai, N., Mimori, T., Yamaguchi-Kabata, Y., Sato, Y., Kawai, Y., & Nagasaki, M. (2014) Lecture Notes in Bioinformatics, 8542, 107–118.
14) Howie, B.N., Donnelly, P., & Marchini, J. (2009) PLoS Genet., 5, e1000529.
15) Sato, Y., Kojima, K., Nariai, N., Yamaguchi-Kabata, Y., Kawai, Y., Takahashi, M., Mimori, T., & Nagasaki, M. (2014) BMC Genomics, 15, 664.
16) Mimori, T., Nariai, N., Kojima, K., Takahashi, M., Ono, A., Sato, Y., Yamaguchi-Kabata, Y., & Nagasaki, M. (2013) BMC Syst. Biol., 7(Suppl 6), S8.
17) Yamaguchi-Kabata, Y., Nariai, N., Kawai, Y., Sato, Y., Kojima, K., Tateno, M., Katsuoka, F., Yasuda, J., Yamamoto, M., & Nagasaki, M. (2015) Human Genome Variation, 2, 15050.
著者紹介Author Profile
 長﨑 正朗(ながさき まさお)
長﨑 正朗(ながさき まさお)東北大学東北メディカル・メガバンク機構ゲノム解析部門バイオメディカル情報解析分野教授.博士(理学).
略歴1976年大阪府に生まれる.98年東京大学理学部情報科学科卒業,2004年同大学院理学系研究科情報科学専攻博士課程修了,05年東京大学医科学研究所ヒトゲノム解析センターDNA情報解析分野助手,その後,07年同助教,11年東京大学医科学研究所ヒトゲノム解析センターゲノム機能解析分野,12年には東北大学東北メディカル・メガバンクゲノム解析部門バイオメディカル情報解析分野教授(現職).
研究テーマと抱負情報科学の立場からライフサイエンスへの貢献を目的としてスーパーコンピュータを用いてビッグデータを自在に解析できるデータサイエンティストを当研究室から育成していきたいです.
ウェブサイトhttp://nagasakilab.csml.org/
趣味散歩.