東北地方は,日本の食料供給基地である.(公財)岩手生物工学研究センターでは,岩手県および東北地方の農業振興を図る目的で,岩手県農業研究センター技術部と密接に連携して,地域に適した水稲優良品種の育成を進めている.その目的で,東北主力品種「ひとめぼれ」を中心材料として,大規模突然変異系統群および交配系統群を育成し,遺伝子資源の充実に努めてきた.これらの遺伝子資源を利用して育種上重要な遺伝子およびゲノム領域を同定して活用する目的で,次世代シークエンサーを用いたイネ全ゲノム解析技術基盤を確立した.本稿では,大規模な水稲遺伝子資源を活用して全ゲノム解析により有用遺伝子領域を迅速に同定し,育種に活用する取り組みについて紹介する.
近年,水稲の育種に対して多様なニーズがあげられている.「コシヒカリ」を超える良食味性,高品質,高温登熟耐性,高度耐病性,直播適性,さらには食味・品質を維持した上での多収性など,求められている形質は高度かつ多様である.こうしたニーズに応える育種を行うためには,多様な形質を生み出す遺伝子資源が必要である.ところが,わが国において近年育成されている水稲品種は「コシヒカリ」ゲノム偏重であり,多様性が小さいことが全ゲノム解析の結果から指摘されている1).このような状況において従来育種に利用されている品種系統をいくら交配しても画期的な形質を持つ品種は誕生しない.加えて,遺伝的多様性が小さいということは,わが国の水稲栽培が病害や気候変動などに対して脆弱であることを示唆する.そこで我々は,地域に適した新しい品種を生み出すため,①新たな変異を創出する,②多様な自然変異を利用する,という二つの戦略で遺伝子資源の拡充を進めてきた.
一つは,新たな変異の創出を目的に作出した突然変異系統群である.東北地域の主要品種「ひとめぼれ」を材料に,ethyl-methanesulfonate(EMS)処理2)により12,000系統の大規模な突然変異系統群を作出し,適切な管理の下で種子(M3~M5世代)を維持している.これは,国内外をみてもイネにおいて最大規模の統一された突然変異系統群であり,かつ,東北地域での栽培に適した唯一のイネ突然変異系統群であるといえる.
EMSを変異源とする突然変異は,トランジション型のDNA点突然変異が主な変異である.次世代シークエンサーによるリシークエンス(re-sequencing)解析から,我々が作出した突然変異系統群では1系統あたり1,000~2,000 SNPsが挿入されていると推察され,これを12,000系統に当てはめると,任意の1 kbのゲノム領域に約40 SNPs検出できることになる.すなわち,理論上12,000系統を探索すると,イネのすべての遺伝子についてSNPがある系統を見つけることができる.従来なかった形質を獲得できる育種のための遺伝子資源としてだけではなく,正遺伝学/逆遺伝学による遺伝子機能解析にも有用な遺伝子資源である2).
もう一つは,多様な自然変異を利用するため,「ひとめぼれ」と世界の多様なイネを交配して作出した約3,000系統のrecombinant inbred lines(RILs)である.イネ・コアコレクション3)などから選んだ22品種系統と「ひとめぼれ」を交配し,22通りの組み合わせのRILs計3,078系統(各組み合わせ22~250系統,組み合わせあたり平均140系統)を維持している.これらの世代は自殖を進めてF7~F9世代に到達しており(2015年現在),ほぼすべてのゲノム領域がホモ接合体で遺伝的に固定された系統である.反復試験や多地点での栽培試験が可能であり,遺伝子型と環境の相互作用解析やエピスタシス解析など利用範囲は広い.
また,我々は大規模な遺伝子資源の整備と同時に,大規模な形質調査,データ管理,種子・DNA管理を効率的に行うためのツールも開発している4).PDAとバーコードやbluetooth通信によるデータ収集・管理は,圃場における大規模系統群の形質調査に必須のツールとして活躍している.
これらの大規模な遺伝子資源を利用して,ニーズに応えうる育種上有用な遺伝子領域を同定し,ゲノム育種により迅速に品種を育成することが課題である.
1)次世代シークエンサー
現在,当センターでは,illumina社の次世代シークエンサーNextSeq500, MiSeqおよびGAIIxを各1台ずつ保有している.現在,主に利用しているのは,NextSeq500とMiSeqの2台である.NextSeq500の特徴は,シークエンスリード(以下リードと略)あたりの費用が安く,必要なリード量を得るのに必要な稼働時間が短いことである.しかし,高精度の長いリード(150 bp以上)の産出には適さない.後述するMutMapやQTL-seq等は,リシークエンス解析の一種であり,特に高品質で長いリードを必要とする解析ではない.そのため,MutMapやQTL-seq等の解析をする際には,我々はNextSeq500を使用している.一方,Miseqは,高精度の長いリードを産出する能力を有する.そのため,高精度なリファレンス配列を構築することを目的としたde novo assemble用リード産出のために主にMiSeqを使用している.ただし,ゲノムサイズが50 Mb以下の生物種におけるリシークエンス解析など,小規模なシークエンスデータのみを必要とする場合は,MiSeqを使用することもある.これらに加えて,沖縄県農業研究センターおよび信州大学遺伝子実験施設と共同研究を実施し,前者が所有するHiSeq2500,後者の所有するMiSeqを随時利用して,大量のシークエンシングを迅速に実施する体制を整えている.
2)データ解析サーバー
当センターで使用している解析サーバーは,IBM社ラック型サーバーSystem x3850 M2(メモリ:208 GB, CPU:48個,OS:オープンソースCentOS version 5)である.これらは,リシークエンス解析にとっては十分な性能を有するが,de novo assembleなどの大量計算を必要とする解析においては,メモリ不足に陥ることもある.その場合には,大学共同利用機関法人国立遺伝学研究所のスーパーコンピューターを共同利用している(国立遺伝学研究所のウェブサイト参照,https://sc.ddbj.nig.ac.jp).また,シークエンスデータ保存用のファイルサーバーとしては,Dell社PowerEdge R620(ハードディスク容量:90 TB, OS:商用RedHat Enterprise Linux version 6)を使用している.ファイルサーバーに関する留意点は,バックアップ体制の整備である.以前,我々も落雷によりデータが一部消失する被害に見舞われた.シークエンスデータのバックアップは必須である.
3)バイオインフォマティクス解析人員
現在,我々のグループでは,各研究者は,研究対象の生物材料のサンプリングまでを担当し,それ以降2名の専属スタッフが,DNA抽出からライブラリー調整,そして,シークエンサーのrunまでを担当している.シークエンスデータ取得後,各研究者は,4名のバイオインフォマティクススタッフとともにデータ解析方法について相談しながら研究を進める体制をとっている.
近年においては,シークエンスだけでなくデータ解析についても外注が可能であるが,我々は,あえてすべての解析を可能な限り自力で行う方針をとっている.有意義かつ効率的なデータ解析のためには,シークエンスする材料,ライブラリー構築,解析手法のすべてについて,詳細な内容を関係者が把握する必要があると考えるからである.さらに,圃場での作業からシークエンス解析結果の吟味までの時間を短くすることが可能であり,効率的なフィードバックが成果につながると判断している.圃場での作業からデータ解析までを担当する10名以上のメンバーが,一丸となって目的とする結果を出すことを心がけている.
前述のとおり,我々が保有する12,000系統の「ひとめぼれ」突然変異系統群は,イネ遺伝子の機能解明,ならびに育種利用を目的とした遺伝子資源として有用性が高い.適切な手法で表現型を観察して突然変異体を得た後,その遺伝子を同定することが遺伝子機能解明やゲノム育種へ向けた第一歩となる.従来は,突然変異の原因遺伝子を単離するために,突然変異体と遠縁の系統とを交配し,その交雑後代を用いて連鎖解析を行うことが定石であった.連鎖解析に必要な多型DNAマーカーを数多く得るため,遠縁の系統,たとえばイネでは二つの亜種であるjaponicaとindicaといった組み合わせで交配する必要があったが,これでは系統間にもともと存在する量的形質遺伝子座(quantitative trait loci:QTL)によって目的の突然変異形質がマスクされてしまう可能性が高い.特に育種に利用したい量的形質の小さな変異ほどこの問題に直面する.
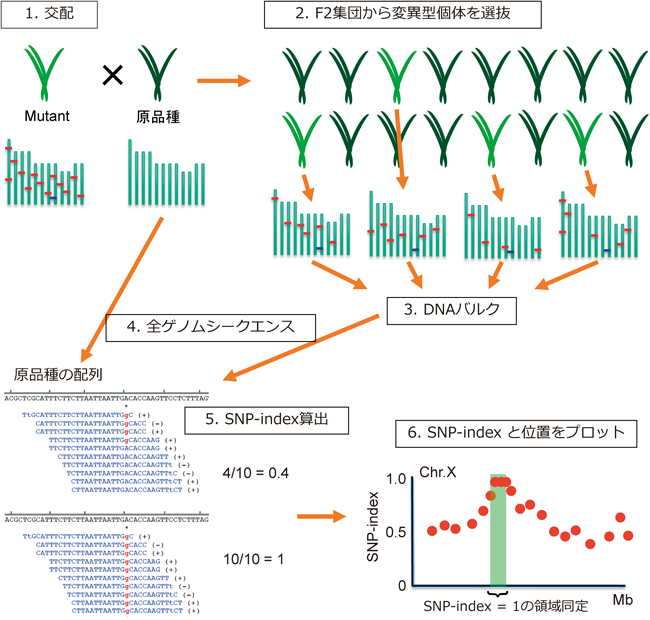
そこで我々は,突然変異形質の原因遺伝子を全ゲノム解析により迅速に同定する技術として,MutMap法を開発した(図1)5).MutMap法では,まず目的の表現型を示す突然変異体と変異処理に用いた原品種とを交配し,得られたF1個体を自殖させ,F2集団を得る.次に,F2集団を栽培し,適切な方法で変異形質を持つ個体を選抜する.仮に,野生型(原品種型)と変異型が3:1の割合で分離した場合,変異形質は一つの劣性突然変異によってもたらされたとわかる.F2集団から変異型を示す約20個体を選抜し,そのゲノムDNAを均等に混合し,次世代シークエンサーにより全ゲノムシークエンシングする(バルクシークエンシング).MutMap法では,20個体程度の変異型個体のゲノムDNAをバルクシークエンシングすることで解析が可能である.すなわち,劣性1遺伝子の突然変異であれば100個体のF2集団を栽培すれば,変異型20個体を選抜することが可能であり,省力化が可能である.
次に,シークエンシングで得られた大量の75~100塩基の塩基配列(リード)を解析する.バルクシークエンシングで得られたリードを,原品種のゲノムDNAをシークエンシングしてあらかじめ作成した基準配列に対してアライメントし,SNP-indexを計算する.SNP-indexとは,基準配列上のある塩基にアライメントされたリード全体のうち,基準配列と異なる塩基を持つリードの割合である.図1の例では,張りついている10リードのうち,4リードが基準配列と異なっているので,SNP-index=4/10=0.4と計算される.F2集団から選抜した突然変異形質を示す20個体は,すべての個体が変異の原因SNPをホモ接合で保有している.すなわち,バルクしたDNAではすべてのリードがSNPを持っていることになり,SNP-index=1が期待される.一方で,突然変異体が保有する変異形質と無関係のSNPは,F2個体のバルクDNAでは,SNPを含むリードと含まないリードが1 : 1,つまりSNP-indexは0.5となることが期待される.このSNP-indexの計算を,突然変異体と原品種との間のすべてのSNPについて行い,横軸をSNPの位置,縦軸をSNP-indexとして染色体ごとにプロット図を作成する.このプロット図からSNP-indexが1となる領域を見いだすことで,変異形質の原因SNPを同定することができる.このビッグデータ解析においては,前述のバイオインフォマティクス環境が必要となる.
MutMap法の利点として次の2点があげられる.一つは,連鎖解析のための多型DNAマーカーが不要なことである.従来は多くの時間と労力を要した遺伝子型判定が,一度の全ゲノムシークエンシングに代替され,迅速な遺伝子単離を可能にしている.もう一つは,量的形質における効果の小さい変異の原因遺伝子を同定できることである.MutMap法では,突然変異体とその原品種を交配するため,交雑後代の表現型を変異型と野生型に明確に区別でき,これまで扱うことが難しかった変異形質であっても解析が可能になったといえる.なお,F3以降での解析や交配ができない変異形質の解析のためMutMap+法6),基準配列にないゲノム領域の変異を同定する手法としてMutMap-Gap法7)が,派生技術として確立されている.
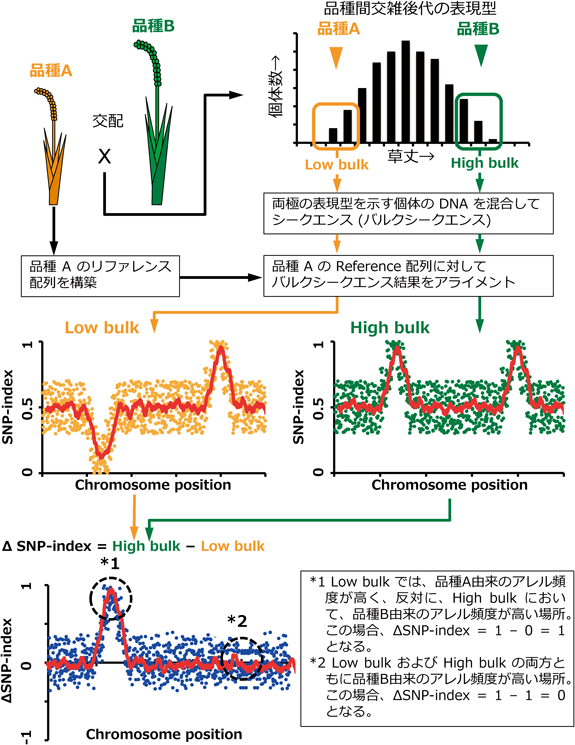
人為的に育成された突然変異体の原因遺伝子を同定する方法が,MutMap法であるのに対して,QTL-seq法は,品種間の形質の違いを決定している量的遺伝子座(QTL)を同定する技術である8).
QTL-seq法の原理は,次のとおりである(図2).まず,品種間の交雑(品種A×品種B)後代において表現型を調査し,対極の表現型を示す個体をそれぞれ20個体以上選抜する.次に選抜した表現型ごとに各個体からDNAを等量ずつ混合してバルクDNAを用意し(Low bulkおよびHigh bulk),次世代シークエンサーによるシークエンシングに供する(バルクシークエンシング).各バルクシークエンシングの結果得られるリードは,それぞれ,交配に用いた片親(例:品種A)のリファレンス配列に対してアライメントされる.アライメント後,前述のSNP-indexを全ゲノムにわたって計算する.バルクシークエンシングにおけるSNP-indexは,リファレンス配列と異なるアレルの頻度を表す値である.それゆえ,QTL-seqの場合,バルクシークエンシングに用いたすべての個体が,リファレンス配列に用いた品種A親のアレルを有する領域では,SNP-index=0を示す.反対に,選抜個体のすべてが,品種B親のアレルを有する領域では,SNP-index=1となる.最後に,バルクサンプル間のSNP-indexを比較するため,SNP-indexの差分であるΔSNP-indexを計算する.その結果,SNP-indexの差が大きい領域,すなわちバルクシークエンシングに供するため選抜されたサンプル間でアレル頻度に大きな差がある領域が,高いΔSNP-index(0<)または,低いΔSNP-index(<0)として検出される.
従来のQTL解析においては,品種間で多型を示すDNAマーカーを設計して,その品種間交雑後代の分離集団において興味のある表現型の分離と連鎖して分離するマーカーを見つけることによりQTLの位置を推定する手法が用いられてきた.そのため,多型マーカーを設計しやすいと考えられる遠縁品種間の交雑後代が解析に利用されてきた.QTL-seqでは,バルクサンプルを全ゲノムシークエンンシングすることで,品種間に存在するすべてのSNP箇所をマーカーとして利用できる.そのため,交配する品種間の類縁関係には制限がない.
6. Nested Association Mapping(NAM)
近年のシークエンシング技術あるいはSNPタイピング技術の発展により,生物のDNA変異をゲノムワイドに調べることが容易になった.このため,従来の交配実験によるQTL解析に替わって,直接的な血縁関係のない多数の遺伝資源について表現型とDNA型を計測し,交配実験を行うことなく表現型に関与する遺伝子座を検出しようとするアソシエーション解析が行われるようになった.この状況はイネにおいても同様であり,多くのアソシエーション解析がさまざまな形質を対象に試みられている.アソシエーション解析は,すでにある遺伝資源を用いるため実験材料作出の必要がない,多くのアレルを扱える,多数のSNPsを用いるため高解像度で解析できるなど利点が多いとされている.しかしながら,特にイネでは,解析材料とする遺伝資源の潜在的な集団構造(連鎖不平衡)によって偽陽性・偽陰性が生じやすいという問題があり,統計的パワーが低いことが指摘されている9).実際,イネにおいてアソシエーション解析によって新規に遺伝子同定に成功した例は多くないように見受けられる.
そこで我々は,Nested Association Mapping(NAM)による遺伝子・有用アレルの同定を試みている.Yuらがトウモロコシを材料に提唱したNAMは,複数のfounder系統(既存品種)と一つの共通親との交雑から得られた複数組み合わせの数千系統のRILsを用いて,ゲノムワイドアソシエーション解析(GWAS)を行う手法である10).NAMは,①founderのゲノムをシャッフルすることで集団構造がキャンセルされ統計的パワーが高いこと,②アソシエーション解析と同様に多数のアレルを解析できること,③数千系統が持つ組換えと多数のSNPsによる高解像度のマッピングができることなど,従来のQTL解析やアソシエーション解析に比べてメリットが大きいとされている10, 11).
すでに,イネNAM集団の確立を進め,22通りの組み合わせ計3,078 RILs(F7~F9)を育成し,そのうち18通りの組み合わせ2,026 RILs(F9)について1,015個のSNPマーカーによる遺伝子型判定を完了している.予備的に,この2,026系統を圃場で栽培して得た出穂期の形質データでGWASを行ったところ,多数のQTL検出に成功した.検出された出穂期のQTLは,すでに報告されている遺伝子と数kb~1 Mbの距離に検出した.すなわち,イネNAM集団で興味ある形質を調べることで,これまで見いだされていないQTL遺伝子を狭い領域に同定できることが示唆される.
現在,確立した約3,000系統のイネNAM集団を用いて,育種上重要な形質に関与する遺伝子の同定を目的に研究を進めている.
我々の目的は,大規模遺伝子資源とそのゲノム情報から育種上重要な形質を支配する遺伝子を迅速に同定するとともに,同定された遺伝子または遺伝子領域に強く連鎖したDNAマーカーを開発して,実際の育種を担っている岩手県農業研究センターにおける効率的な交配育種(DNAマーカー育種,ゲノム育種)を支援することにある.ここで,ゲノム育種によって育成した品種の一例を示す.
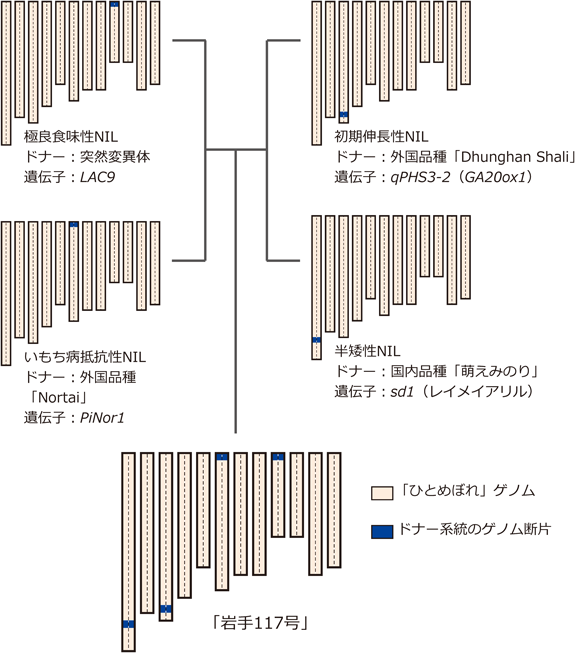
1)有用形質を集積させた水稲品種「岩手117号」
宮城県が育成した水稲品種「ひとめぼれ」は,穂ばらみ期の耐冷性が強く,玄米品質がよく良食味であるなど優れた特性から東北地域の主要品種となっている.しかし,いもち病抵抗性が弱い,耐倒伏性が弱いなど欠点もある.さらに,近年ではさらなる良食味化など形質の高度化が求められ,減農薬栽培や代掻きした水田に直接種子を播く直播栽培が普及するなど栽培方法も多様化している.そこで,極良食味性,いもち病抵抗性,直播栽培に適した初期伸長性,耐倒伏性を向上させる半矮性の四つの形質を「ひとめぼれ」ゲノムに集積させるゲノム育種を,岩手県農業研究センターと共同で実施した(図3).
従来にはない極良食味性を実現するため,「ひとめぼれ」突然変異系統群から白米中のデンプンの一種アミロースの含量がわずかに低くなる変異系統の探索を行った.見いだした変異系統についてMutMap法を適用し,遺伝子を同定することでDNAマーカー選抜を可能とした.いもち病抵抗性は,「ひとめぼれ」といもち病抵抗性を示す外国品種「Nortai」との交配から得られたRILsを用いて,QTL-seq法によりいもち病抵抗性に関与する遺伝子領域を同定した.初期伸長性は従来のQTL解析で同定した遺伝子領域qPHS3-212)を,耐倒伏性は既知の半矮性遺伝子sd113)を利用した.
まず,上記4形質について,遺伝子本体または遺伝子の極近傍にDNAマーカーを設定し,それぞれの形質を単独に持つ準同質遺伝子系統(near isogenic line:NIL)を4系統作出した.NILの作出においては,「ひとめぼれ」を反復親とする戻し交配の過程でゲノムワイドに遺伝子型を確認し,「ひとめぼれ」ゲノムの割合が高い個体を迅速に選抜した.次に,極良食味性NILといもち病抵抗性NIL,初期伸長性NILと半矮性NILの二つの組み合わせでNILどうしを交配し,DNAマーカーで二つの形質を持つ個体を選抜した.さらに,その個体どうしを交配し,DNAマーカーで4形質を持つ個体を選抜することで,「ひとめぼれ」ゲノムに四つの遺伝子を集積させた系統「岩手117号」を短期間で育成することに成功した(図3).
現在,集積した遺伝子領域の効果確認や栽培特性の調査を進めているところであるが,さらなる形質の集積に向けた遺伝子同定とNIL作出も着々と進んでいる.
2)耐塩性水稲品種「Kaijin」
2011年3月11日,東日本大震災に伴う巨大津波および地盤沈下により,東北太平洋沿岸地域の水田には大規模な塩害が生じた.この事態を受けて,我々のグループでは,塩害田においても作付け可能な耐塩性品種を育成するプロジェクトを立ち上げた.最終的な目標にしたのは,その地域で栽培されている系統と同等の栽培特性を持ち,かつ耐塩性が付与された系統,つまり耐塩性の高い「ひとめぼれ」の育成である.
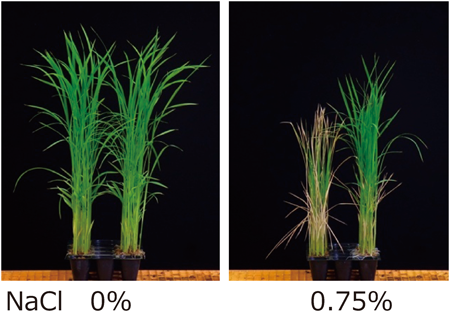
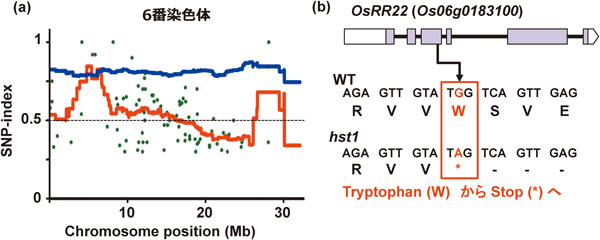
上記目的のため,前述の「ひとめぼれ」突然変異系統群の中から耐塩性変異体をスクリーニングした.約6,000系統の変異体を50%に希釈した人工海水に浸漬した結果(図4),高い耐塩性を示す個体(以下,hst1とする)を選抜することに成功した.幼苗および圃場における試験では,hst1は,「ひとめぼれ」が枯死する塩濃度においても生育した(図5,図6).
hst1変異体に対してMutMap法を適用することより,hst1の原因突然変異箇所が,6番染色体に座乗するB-type response regulatorをコードする遺伝子Os06g0183100のナンセンス突然変異であることを特定した(図7)14).変異体同定からわずか1年で,遺伝子同定に至った.
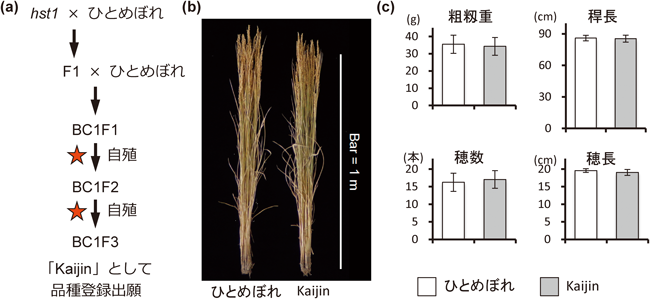
次に,連続戻し交配により,hst1が有する不必要な突然変異箇所を除いた.前述のとおり,選抜された「ひとめぼれ」突然変異体は,EMS処理によりゲノム上の約1,000~2,000か所にランダムに塩基置換が生じている.これらの変異により,不良形質が生じる可能性も危惧されたことから,hst1に「ひとめぼれ」を連続戻し交配することにより,Os06g0183100における耐塩性に関与するSNP以外の箇所をできる限り原品種「ひとめぼれ」に戻す作業を実施した.その際,hst1の原因変異箇所にDNAマーカーを設定し,hst1変異を遺伝している個体のみ選抜し戻し交配に用いた.以上の過程を経て,高い耐塩性を有する「ひとめぼれ」を育成し,「Kaijin」と命名した.通常の栽培環境においては「Kaijin」の主要栽培形質は,「ひとめぼれ」とほぼ同等であり,特に目立った不良形質は観察されないことから,本品種は実用性の高い耐塩性品種と考えられる.「Kaijin」は,現在品種登録申請中である(図8).現在,「Kaijin」の耐塩性メカニズムの解明に取り組むと同時に,実際の現場において「Kaijin」の実用性試験に取り組んでいる.
次世代シークエンサーによる全ゲノム解析が多くの生物で可能になったことにより,形質を支配する遺伝子を迅速に同定することが可能となった.「岩手117号」の例にあるように,有用遺伝子を交配によって集積した新品種の「ゲノム育種」が可能となった.耐塩性系統「Kaijin」で示したように,緊急の必要に応じて新品種を数年で育成することも可能となった.同様の手法により,今後さまざまな作物で,産地および需要に適した品種の迅速な育成が可能になると期待される.
食料生産は,医療と同様,人類の死活に関わる問題である.気候変動により,世界で食料問題が頻発しており,ますます品種育成の重要性が増している.本項で紹介した「ゲノム育種」が,こうした問題に取り組む上で果たす役割は大きいものと期待している.
謝辞Acknowledgments
本研究は,農林水産業・食品産業科学技術研究推進事業の支援を受けて実施した.本研究を支援してくださった(公社)農林水産・食品産業技術振興協会(JATAFF)の黒田秧博士に感謝いたします.
引用文献References
1) Yamamoto, T., Nagasaki, H., Yonemaru, J., Ebana, K., Nakajima, M., Shibaya, T., & Yano, M. (2010) BMC Genomics, 11, 267.
2) Rakshit, S., Kanzaki, H., Matsumura, H., Rakshit, A., Fujibe, T., Okuyama, Y., Yoshida, K., Tamiru, M., Shenton, M., Utsushi, H., Mitsuoka, C., Abe, A., Kiuchi, Y., & Terauchi, R. (2010) In The Handbook of Plant Mutation Screening (Meksem, K. & Kahl, G. eds.), pp. 187–197, WILEY-VCH.
3) Kojima, Y., Ebana, K., Fukuoka, S., Nagamine, T., & Kawase, M. (2005) Breed. Sci., 55, 431–440.
4) Utsushi, H., Abe, A., Tamiru, M., Ogasawara, Y., Obara, T., Sato, E., Ochiai, Y., Terauchi, R., & Takagi, H. (2015) Breed. Sci., 65, 285–289.
5) Abe, A., Kosugi, S., Yoshida, K., Natsume, S., Takagi, H., Kanzaki, H., Matsumura, H., Yoshida, K., Mitsuoka, C., Tamiru, M., Innan, H., Cano, L., Kamoun, S., & Terauchi, R. (2012) Nat. Biotechnol., 30, 174–178.
6) Fekih, R., Takagi, H., Tamiru, M., Abe, A., Natsume, S., Yaegashi, H., Sharma, S., Kanzaki, H., Matsumura, H., Saitoh, H., Mitsuoka, C., Utsushi, H., Uemura, A., Kanzaki, E., Kosugi, S., Yoshida, K., & Terauchi, R. (2013) PLoS ONE, 8, e68529.
7) Takagi, H., Uemura, A., Yaegashi, H., Tamiru, M., Abe, A., Mitsuoka, C., Utsushi, H., Natsume, S., Kanzaki, H., Matsumura, H., Saitoh, H., Yoshida, K., Cano, L., Kamoun, S., & Terauchi, R. (2013) New Phytol., 200, 276–283.
8) Takagi, H., Abe, A., Yoshida, K., Kosugi, S., Natsume, S., Mitsuoka, C., Uemura, A., Utsushi, H., Tamiru, M., Takuno, S., Innan, H., Cano, L., Kamoun, S., & Terauchi, R. (2013) Plant J., 74, 174–183.
9) Han, B. & Huang, X. (2013) Curr. Opin. Plant Biol., 16, 133–138.
10) Yu, J., Holland, J.B., McMullen, M.D., & Buckler, E.S. (2008) Genetics, 178, 539–551.
11) Tian, F., Bradbury, P.J., Brown, P.J., Hung, H., Sun, Q., Flint-Garcia, S., Rocheford, T.R., McMullen, M.D., Holland, J.B., & Buckler, E.S. (2011) Nat. Genet., 43, 159–162.
12) Abe, A., Takagi, H., Fujibe, T., Aya, K., Kojima, M., Sakakibara, H., Uemura, A., Matsuoka, M., & Terauchi, R. (2012) Theor. Appl. Genet., 125, 647–657.
13) Sasaki, A., Ashikari, M., Ueguchi-Tanaka, M., Itoh, H., Nishimura, A., Swapan, D., Ishiyama, K., Saito, T., Kobayashi, M., Khush, G.S., Kitano, H., & Matsuoka, M. (2002) Nature, 416, 701–702.
14) Takagi, H., Tamiru, M., Abe, A., Yoshida, K., Uemura, A., Yaegashi, H., Obara, T., Oikawa, K., Utsushi, H., Kanzaki, E., Mitsuoka, C., Natsume, S., Kosugi, S., Kanzaki, H., Matsumura, H., Urasaki, N., Kamoun, S., & Terauchi, R. (2015) Nat. Biotechnol., 33, 445–449.
著者紹介Author Profile
 阿部 陽(あべ あきら)
阿部 陽(あべ あきら)(公財)岩手生物工学研究センター研究主査.博士(農学).
略歴1975年青森県に生る.2001年岩手大学大学院農学研究科修了.12年同大学院連合農学研究科修了.02年岩手県農業研究センター研究員,12年より現職.
研究テーマと抱負イネ重要形質の遺伝子単離と機能解析.作物の画期的品種の開発に貢献.
ウェブサイトhttp://genome.ibrc.or.jp
高木 宏樹(たかぎ ひろき)(公財)岩手生物工学研究センター主任研究員.博士(農学).
略歴1984年富山県に生る.2007年新潟大学農学部卒業.09年新潟大学大学院自然科学研究科修了.10年岩手生物工学研究センター入社.14年岩手大学大学院連合農学研究科にて博士号取得.同年より現職.
研究テーマと抱負本稿で紹介したMutMapやQTL-seqなどの次世代シーケンサーを用いた新規遺伝子単離技術の開発を行っている.また,開発された技術を用いて,イネにおける栽培上有用な遺伝子の同定も挑戦している.
ウェブサイトhttp://genome.ibrc.or.jp
趣味山へ花を見に行くこと.街へ人を見に行くこと.
夏目 俊(なつめ さとし)(公財)岩手生物工学研究センターゲノム育種研究部研究専門員.
略歴1992年岩手大学人文社会科学部人文社会科学科卒業.IT業界に就職し宮城県で起業,岩手県で企業活動を行う.現在は岩手生物工学研究センターにてバイオインフォマティシャンとして活動の傍ら,博士課程で研究を行なっている.
研究テーマと抱負雌雄異株の野生草本ヤマノイモ属オニドコロを中心として,デノボ・アセンブリ技術を用いて非モデル植物のドラフトゲノム構築を最適化する研究を行っている.
趣味コントラクトブリッジ.
八重樫 弘樹(やえがし ひろき)(公財)岩手生物工学研究センターゲノム育種研究部.
略歴1990年東北大学工学部卒業.90年ソニー株式会社.11年より現職.
研究テーマと抱負次世代シーケンサを用いたゲノム解析技術の開発とその応用.
趣味スポーツ観戦.
菊池 秀子(きくち ひでこ)(公財)岩手生物工学研究センター契約技術職員.
ウェブサイトhttp://genome.ihrc.or.jp
趣味マラソン.
吉田 健太郎(よしだ けんたろう)神戸大学自然科学系先端融合研究環助教.博士(農学).
略歴1977年島根県に生る.2001年京都大学農学部卒業.06年に京都大学農学研究科において博士(農学)を取得.06年から12年まで財団法人岩手生物工学研究センターで研究員.12年から14年までイギリスセインズベリー研究所で研究員.14年から現職.
研究テーマと抱負研究テーマは,植物と病原菌の相互作用進化のメカニズムを解明することです.野外環境下において予想を超えるようなダイナミックな植物と病原菌の分子レベルでの攻防を明らかにしていきたいです.
ウェブサイトhttp://www.lab.kobe-u.ac.jp/ans-plantgenetics/index.html
趣味スキー,ハイキング.
小杉 俊一(こすぎ しゅんいち)理化学研究所統合生命医科学研究センター研究員.博士(農学).
略歴筑波大学生物学類卒業.同大学院農学研究科博士課程修了後,生物資源研究所,慶應義塾大学,岩手生物工学センター等を経て,現職に至る.
研究テーマと抱負NGSを用いたゲノム情報解析・解析ツールの開発,およびタンパク質細胞内移行制御に関する解析.
齋藤 宏昌(さいとう ひろまさ)(公財)岩手生物工学研究センター主任研究員.博士(農学).
略歴1993年帝京大学理工学部卒業.95年三重大学大学院生物資源学研究科博士前期課程修了.98年大阪府立大学大学院農学研究科博士後期課程単位修得退学.99年博士(農学)取得(大阪府立大学).98~2002年財団法人岩手生物工学研究センター研究員.03~05年Max Planck Institute for Plant Breeding Research(ドイツ)ポスドク研究員.05~10年財団法人岩手生物工学研究センターポスドク研究員.10年より現職.
研究テーマイネ—いもち病菌相互作用の解明.
抱負エフェクター分泌によるいもち病菌のイネへの感染および抵抗性誘導機構を解明したい.
ウェブサイトhttp://genome.ibrc.or.jp/
趣味バレーボール,テニス.
浦崎 直也(うらさき なおや)沖縄県農業研究センター上席主任研究員.博士(農学,2003年鹿児島大学).
略歴1968年沖縄県に生る.97年鹿児島大学大学院連合農学研究科単位取得退学,同年沖縄県農業試験場研究員.10年より現職.
研究テーマと抱負次世代シークエンサーを活用して地域特産作物の育種支援技術を開発し,新品種育成の効率化を目指している.
ウェブサイトhttp://www. pref.okinawa.jp/arc/
趣味マンゴー,パインアップル,ニガウリ,黒糖の食べ比べ.
松村 英生(まつむら ひでお)信州大学ヒト環境科学研究支援センター遺伝子実験部門准教授.博士(農学).
略歴1969年東京都に生まれる.91年東京大学農学部卒業.96年同大学大学院修了.96年より東京大学分子細胞生物学研究所,98年より(財)岩手生物工学研究センターにて研究に従事し,2010年より現職.
研究テーマと抱負遺伝子発現やゲノム多型などの解析に関わる様々な技術改良を行うとともに,それらゲノム解析技術を駆使して各種植物の性決定機構の解明を目指している.
ウェブサイトhttp:// gene_rc.shinshu-u.ac.jp
寺内 良平(てらうち りょうへい)(公財)岩手生物工学研究センターゲノム育種研究部部長.農学博士.