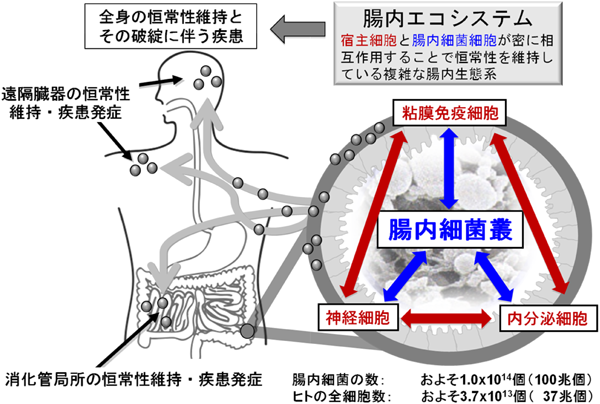
3. 腸内エコシステムの破綻がもたらす疾患と腸内細菌叢由来代謝物質
1)腸管関連疾患
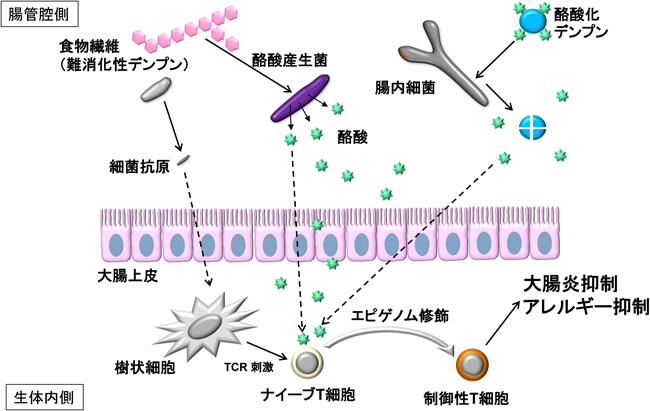
腸内エコシステムのバランスの破綻と腸管関連疾患については,特に慢性炎症疾患と腸内細菌叢との関係について近年多数の報告がある.たとえば,生体の遺伝子異常と腸内エコシステムの破綻がその起因となることが報告されている炎症性腸疾患(inflammatory bowel disease:IBD)では,生体側の遺伝的素因として,これまでに多数の一塩基多型(single nucleotide polymorphism:SNP)が報告されている11).しかしながら,マウスモデルにおいて生体の遺伝子異常のみでは腸炎は自然発症しないことから,腸内細菌叢やウイルス感染などの環境要因の関与も腸炎発症には重要であることが示唆されている.事実,健常者とIBD患者における腸管内容物や腸粘膜における細菌叢構成の相違が報告されており,IBD患者では特に主要な腸内細菌群の一つであるクロストリジウム目細菌群が減少していることが示唆されている12, 13).また,IBD患者の腸内細菌叢はその多様性が低下するが,逆にバクテリオファージの多様性は増加することも報告されている14).クロストリジウム目細菌群は腸管粘膜において免疫応答の抑制に寄与する制御性T細胞(regulatory T cell:Treg細胞)の分化誘導を促すことが報告されていることから,こういった腸内細菌叢の変化がIBD発症と深く関係すると考えられる15, 16).筆者らは,クロストリジウム目細菌群による大腸局所におけるTreg細胞誘導メカニズムについてメタボロゲノミクスにより解析し,クロストリジウム目細菌群が腸管内において,食物繊維の嫌気発酵により産生する短鎖脂肪酸(short-chain fatty acid:SCFA)の一つである酪酸が,大腸粘膜においてナイーブT細胞からTreg細胞をエピゲノム修飾により分化誘導することで,大腸炎の抑制に寄与することを明らかにした(詳細は後述)17).
IBDを含む慢性大腸炎患者がその後大腸がんを発症しやすいことは以前より知られていたが,慢性炎症から大腸がんに移行する際には特定の大腸菌が産生するコリバクチンと呼ばれるポリケチドの一種が,腸管細胞のDNAに傷害を与えることがその原因の一つになることが示唆されている18).他にも,がんと腸内細菌叢由来代謝物質については,古くから腸内細菌が胆汁酸代謝により産生する二次胆汁酸が大腸がん発症と関連があることが知られていたが,7,12-ジメチルベンズ[a]アントラセン(7,12-dimethylbenz[a]anthracene:DMBA)を用いた発がん誘導マウスモデルの血清メタボローム解析により,高脂肪食摂食時に腸管内へと分泌される胆汁中のコール酸が腸内細菌によりデオキシコール酸(deoxycholic acid:DCA)に代謝され,この二次胆汁酸であるDCAが腸管から再吸収された後に肝臓の肝星細胞の老化を促進することで炎症性サイトカインなどの分泌を促し,最終的には肥満に伴って発症する肝臓がんを誘発することも報告された19).腸内細菌叢の16S rRNA遺伝子群のメタゲノム解析から,高脂肪食摂取時にクロストリジウム属細菌群の中のクラスターXIに属する菌種が特徴的に増加し,16S rRNA遺伝子配列の系統樹解析から,最も近縁であったClostridium sordelliiはDCA産生能を有したことから,血清メタボロミクスと腸内細菌叢メタゲノミクスとを組み合わせたメタボロゲノミクスにより,標的であるDCA産生腸内細菌種を同定するに至った.
他にも,種々の遺伝子改変大腸がん発症マウスモデルのメタボロゲノミクスにより,宿主遺伝子異常と腸内細菌叢由来代謝物質との関係が明らかになりつつある.家族性大腸腺腫症の原因遺伝子であるadenomatous polyposis coli(Apc)の遺伝子変異(ApcMin/+)とDNAミスマッチ修復に関わるMutS homolog 2(Msh2)の遺伝子欠損(Msh2−/−)の両方を有するApcMin/+Msh2−/−マウスは大腸がんを発症するが,そのメカニズムとして,腸内細菌叢の発酵代謝により産生される酪酸が大腸上皮細胞の増殖を促すため,結果としてApcMin/+Msh2−/−マウスの大腸上皮細胞では遺伝子変異が蓄積してしまい,大腸がんを発症することが報告されている20).腸内細菌叢の16S rRNA遺伝子群のメタゲノム解析からApcMin/+Msh2−/−マウスではクロストリジウム目細菌群が増加しており,抗生物質投与により腸内細菌叢を除去すると大腸がん発症も有意に抑制されることが明らかとなっている20).また,腸管上皮細胞特異的にがん原遺伝子の一つであるK-ras遺伝子の活性化型変異(K-rasG12Dint)を発現させたマウスへ高脂肪食を摂食させると小腸に腫瘍が形成されるが,これは高脂肪食摂取による肥満とは無関係であり,高脂肪食摂取による小腸内細菌叢の乱れ(dysbiosis)が関与していることが報告されている21).K-rasG12Dintマウスでは対照群と比較してパネート細胞からの抗菌物質産生量やゴブレット細胞からのムチン産生量が低下しており,そのことが高脂肪食摂取による小腸内細菌叢のdysbiosisを促し,腫瘍形成に寄与していると考えられる.事実,抗生物質投与により小腸での腫瘍形成が完全に阻害されることや,酪酸の経口投与によりムチン産生量の回復に伴った小腸内細菌叢の改善が,腫瘍形成を抑制することも報告されている21).
機能性消化管障害に分類され,便通異常や腹痛を伴う過敏性腸症候群(irritable bowel syndrome:IBS)の発症も腸内エコシステムの破綻と関係する可能性が示唆されている.IBSの発症原因はこれまでにストレスによる過度の緊張や精神的不安があげられてきていたが,近年ではストレスによって生じる腸内細菌叢の変化が粘膜局所における微弱な炎症を誘発し,その結果消化管の蠕動運動をつかさどる自律神経系の異常や消化管知覚閾値の低下を伴うイオン輸送能および粘膜透過性の上昇を誘発することが報告されている22).腸内細菌叢の刺激によって腸管内分泌細胞から産生されるセロトニンや,免疫細胞から産生される副腎皮質刺激ホルモン放出ホルモンが消化管蠕動運動に重要な役割を果たしていると考えられている.近年の報告で,芽胞形成性の腸内細菌が腸管内で産生する二次胆汁酸をはじめとするいくつかの代謝物質により,腸管の内分泌細胞からのセロトニン産生が促され,血小板機能や腸管蠕動運動の改善がもたらされることが報告されている23).しかしこれらとIBSとの関連は不明であるため,今後の研究進展が期待される.
2)全身性疾患
近年増加の一途をたどっている肥満や糖尿病,動脈硬化といった全身性の代謝疾患や,その患者数が成人の8人に1人といわれている慢性腎臓病についても腸内エコシステムの乱れが深く関与していることが報告されている3, 24).レプチン遺伝子が変異した肥満マウス(ob/obマウス)の腸内細菌叢を,無菌環境下で飼育した無菌の野生型マウスに定着させると,野生型マウスの腸内細菌叢を定着させた場合よりも有意に体重が増加することが明らかとなった25).一方,ob/obマウスに抗生物質を投与することで腸内細菌叢を除菌すると,抗生物質未処理のob/obマウスと比較して肝脂肪や血漿LPS濃度が低下し,肝臓のグリコーゲン量や血漿のアディポネクチン濃度が増加したことから,抗生物質による腸内細菌叢の除菌はob/obマウスの耐糖能の改善に効果があることが示唆された26).欧米人の双子の腸内細菌叢を用いた研究も実施されており,双子のうち一方が肥満型,もう一方がやせ型のペアのヒト腸内細菌叢を無菌マウスにそれぞれ定着させて通常飼料で飼育した場合,マウスはそれぞれのドナーの体型を反映することが明らかとなった27).興味深いことに,これらのマウスに低脂肪・高繊維食を与えた場合,やせ型腸内細菌叢を定着させたマウスはそのままの表現型だったが,肥満型腸内細菌叢を定着させたマウスはやせ型の腸内細菌叢に近づき,体型もやせる傾向にあることが明らかとなった27).このことから,宿主の代謝表現型は腸内細菌叢と食事との組み合わせによって制御可能である可能性が示唆された.
この他にも,肥満モデルマウスや肥満症のヒトの腸内細菌叢解析から,正常な腸内細菌叢と比べてその細菌数が100~1000分の1に減少している腸内細菌としてAkkermansia muciniphilaが報告されている28).A. muciniphilaをマウスに経口投与することで,肥満に伴って生じるインスリン抵抗性や脂肪の蓄積といったメタボリックシンドロームの症状を改善できることが示唆されたことから,本菌はヒトにおいても代謝疾患の予防や治療に有用な腸内細菌である可能性がある29).ヒトの遺伝的素因に関連する腸内細菌とBMI値との関係についての報告もあり,416組の双子を含む1,000以上の便サンプルのメタゲノム解析から,Christensenellaceae科細菌の存在量は二卵性双生児どうしと比べて一卵性双生児どうしで相関しており,またその存在量は低BMI値と正の相関があることが報告されている.実際に,肥満症の人の腸内細菌叢を定着させたマウスへ,Christensenellaceae科細菌の一種であるChristensenella minutaを投与したところ,体重増加の抑止に寄与することが明らかとなった30).中国人の2型糖尿病患者における腸内細菌叢のメタゲノム研究も報告されており,当該論文の著者らが確立したmetagenome-wide association study(MGWAS)により対照群との比較解析から見いだした50個の腸内細菌マーカー遺伝子群に基づいて,2型糖尿病発症リスクが判別可能であることも報告されている31).同様の2型糖尿病コホート研究がヨーロッパでも実施されており,興味深いことにヨーロッパ人の2型糖尿病発症リスク判別因子は中国人のコホート研究で見いだした50個の腸内細菌マーカー遺伝子群とは異なっていた32).このことから,腸内細菌叢プロファイルに基づく2型糖尿病発症リスクの予測モデルを構築するには,人種や年齢などの宿主因子や地域差といった環境要因も考慮する必要が示唆された.
食事の欧米化が生活習慣病のリスクを高めていることも以前より知られているが,レシチンなどの食事由来のコリンや赤身肉中に多く含まれるL-カルニチンを摂取すると腸内細菌によりトリメチルアミン(trimethylamine:TMA)に代謝され,消化管から吸収されたTMAがさらに肝臓でトリメチルアミン-N-オキシド(trimethylamine N-oxide:TMAO)に代謝されることで,アテローム性動脈硬化を促進することが,マウスを用いた実験やヒト血清のメタボローム解析より明らかとなっている33, 34).また近年その使用が増加しているサッカリンなどの人工甘味料が腸内環境に与える影響も検討されており,サッカリンの摂取が腸内細菌叢の変化を介して耐糖能の悪化を招くことがマウスを用いた実験により報告されている35).ヒトにおいては人工甘味料を長期的に摂取している人は血糖値が高い傾向があることが示唆されており,人工甘味料の摂取試験では,耐糖能が悪化する人(レスポンダー)と,悪化しない人(非レスポンダー)が存在し,その違いはもともとの腸内細菌叢プロファイルに起因することも明らかとなっている35).食品添加物の一種である乳化剤の摂取についても腸内環境に与える影響が検討されており,マウスモデルにおいて乳化剤の摂取が腸内細菌叢の変化を介して大腸炎の発症を促すことや,肥満や血糖値上昇といったメタボリックシンドローム症状を引き起こすことも報告されている36).
精神疾患の一つである自閉症スペクトラム障害の発症に腸内細菌叢の乱れが関与することが報告されている.妊娠マウスにpoly(I : C)を腹腔内投与し母体免疫を活性化することで,その産仔が自閉症様症状を呈するモデルマウスにおいて,腸内細菌叢の16S rRNA遺伝子群のメタゲノム解析および血清のメタボローム解析を行ったところ,仔マウスの腸内細菌叢の乱れ(dysbiosis)が腸管バリア機能の低下を招き,その結果腸内細菌叢から産生される尿毒症物質の一つである4-ethylphenylsulfateが腸管内から血中に移行することで自閉症様症状を呈することが明らかとなった37).このとき,腸管バリア機能を改善するようなBacteroides fragilisをプロバイオティクスのように投与すると血中の4-ethylphenylsulfate濃度が低下し,それに伴って自閉症様症状も改善されたことから,プロバイオティクス摂取による腸内エコシステムの改善が,自閉症治療のターゲットの一つとなりうる可能性が示唆された.
慢性腎臓病と腸内細菌叢との関連も報告されており,腸内細菌叢から産生される種々の尿毒症物質が慢性腎臓病の発症や増悪に関わる可能性が示唆されている38).筆者らもアデニン食摂食による腎不全発症マウスモデルにおいて,塩化物イオンチャネル活性化剤で慢性便秘治療に用いられるルビプロストンの経口摂取により腸内環境を改善すると,腎不全の病態スコアを改善できることをメタボロゲノミクスにより明らかにした.腎不全発症による腸内細菌叢の乱れがルビプロストン摂取により改善され,その結果血中のインドキシル硫酸や馬尿酸といった腸内細菌叢由来の尿毒症物質濃度が低下したことが,腎不全病態スコアの改善につながったと考えられる39).
自己免疫疾患の一つである多発性硬化症と腸内細菌叢との関係についても報告があり,多発性硬化症のマウスモデルである実験的自己免疫性脳脊髄炎(experimental autoimmune encephalomyelitis:EAE)マウスに非吸収性の抗生物質を摂取させることで腸内細菌叢構成が変化し,その結果EAEの臨床スコアが有意に低下することが報告されている40).同様に多発性硬化症患者の腸内細菌叢についても検討されており,腸内細菌叢の多様性は健常者と同程度だったものの,患者内の腸内細菌叢のばらつきは,健常者のそれよりも大きかったことから,中程度の腸内細菌叢構造異常(dysbiosis)が生じている可能性が示唆された41).特にクロストリジウム属細菌群の中のクラスターIVやXIVaに属する細菌群が多発性硬化症患者群で減少しており,これらの菌群はTreg細胞の分化誘導に関与することが明らかとなっていることから15, 16),これらの腸内細菌叢変化と病態発症との関連についてさらなる研究の進展が期待される.
4. 腸内細菌叢由来代謝物質がもたらす生体恒常性維持機構
1)粘膜免疫システムと腸内細菌叢由来代謝物質
腸内エコシステムの恒常性維持において,腸内細菌叢がもたらす多様な機能のうち,その多くは粘膜免疫システムの制御に関わっていることが近年明らかになりつつある.腸内細菌叢を持たない無菌マウスでは,CD4 T細胞およびインターロイキン-4(interleukin-4:IL-4)依存的に,B細胞から免疫グロブリンE(immunoglobulin E:IgE)産生細胞へのクラススイッチが生じることから,無菌マウスの血中IgEレベルは高いことが知られている.しかし,生後直後から多様性の高い腸内細菌叢にさらされることで,B細胞がIgE産生細胞ではなく免疫グロブリンA(immunoglobulin A:IgA)産生細胞へとクラススイッチする結果として,マウス血中IgEレベルは減少し,アレルギーのリスクが低下することが報告されている42).興味深いことに,多様性の不十分な腸内細菌叢の定着では血中IgEレベルが高いままだったことから,生後早い段階で多様性の高い正常な腸内細菌叢が腸管内に定着することが,その後のアレルギー発症リスクを低下させるのに重要であることが示唆され,これは衛生仮説を腸内細菌叢の観点から支持する結果と考えられる42).
腸内細菌叢と免疫細胞の分化に関する報告として,腸内細菌の一種であり,難培養性細菌として知られている腸管セグメント細菌(segmented filamentous bacterium:SFB)は,粘膜免疫系においてIgA産生細胞や腸管上皮細胞間リンパ球(intraepithelial lymphocyte:IEL)を誘導するだけでなく43),宿主免疫との相互作用を介して,宿主防御反応や自己免疫性疾患に関与するインターロイキン-17(IL-17)産生性のヘルパーT細胞(TH17細胞)の集積を促すことが報告されている44–46).難培養性であるSFBの培養方法が近年確立されたことから47),TH17細胞の分化誘導に関わるSFB側因子の同定に今後つながることを期待したい.
主要な腸内細菌群の一種であるクロストリジウム目細菌群が,前述のとおり免疫応答の抑制に重要な役割を担うTreg細胞の分化・誘導を促すことも報告されている15, 16).この効果は特に,クロストリジウム目細菌群のクラスターIVやXIVaに属する細菌群に認められているものの,これらの腸内細菌が有するどのような因子が宿主細胞にどのように作用することで,免疫細胞の分化・誘導に寄与しているのか,すなわち宿主免疫システム-腸内細菌間クロストークの分子機構は不明であった.そこで筆者らは,特にクロストリジウム目細菌群によるTreg細胞の分化誘導メカニズムに焦点を当て,メタボロゲノミクスにより,その分子機構の解明を試みた.
SPF(specific pathogen-free)マウスの腸内細菌叢をクロロホルム溶液により処理し,その懸濁液を無菌マウスに投与することで,クロロホルム耐性菌(chloroform-resistant bacteria:CRB)定着マウス(CRBマウス)を作製した15).次世代シークエンサーを用いた腸内細菌叢の16S rRNA遺伝子群のメタゲノム解析により,CRBマウス腸内細菌叢のうち96.6%はクロストリジウム目の細菌群で占められていることを確認した.本マウスでは無菌マウスと比較して大腸粘膜における末梢誘導型のTreg細胞(peripherally derived Treg cell:pTreg細胞)の数が著しく増加していた17).無菌マウスでは腸内発酵が起こらないため,食物繊維が盲腸内で滞留し水分を含んで膨潤するため盲腸組織が肥大化する現象が観察されるが,CRBマウスではその盲腸肥大が正常化していたことから,クロストリジウム目細菌群が食物繊維を代謝して腸内発酵を促進し,その過程で産生される多様な代謝物質が,大腸粘膜におけるpTreg細胞の誘導に重要な役割を果たしているのではないかと推測した.これを検証するため,CRBマウスに高繊維食または低繊維食を与えることでCRBマウス腸内細菌叢による代謝変動を促した.その結果,腸内発酵があまり起こらない低繊維食群では,腸内発酵が活発な高繊維食群と比較して,大腸のpTreg細胞数が半減していた17).このことから,クロストリジウム目細菌群由来の発酵代謝物質がpTreg細胞誘導に主要な役割を果たすことが明らかとなった.
次に高繊維食および低繊維食を与えたCRBマウス盲腸内容物のメタボロミクスにより,酢酸やプロピオン酸,酪酸などの短鎖脂肪酸や,ロイシンやイソロイシン,γ-アミノ酪酸といったアミノ酸の含量が,高繊維食を与えたCRBマウスの盲腸内で増加していることが明らかとなった.これらの代謝物質のTreg細胞誘導作用を調べるため,in vitro試験系においてTreg細胞分化誘導条件下でナイーブT細胞を培養する際に,盲腸内容物のメタボロミクスにより同定された各代謝物質を添加して共培養し,そのTreg細胞誘導作用を評価した.その結果,酢酸およびアミノ酸にはTreg細胞への分化誘導作用はまったく認められなかったが,プロピオン酸にはわずかな,酪酸には顕著なTreg細胞分化誘導作用が認められた17).酪酸によるTreg細胞誘導能を個体レベルでも評価するため,酪酸をデンプンに架橋した酪酸化デンプンをSPFマウスに与え,腸管内での酪酸量を増加させたところ,大腸粘膜におけるpTreg細胞の数が対照群と比較しておよそ2倍にまで増加した.このとき,卵白アルブミンに特異的なT細胞抗原受容体を発現させたトランスジェニックマウス(OT-IIマウス)由来のナイーブT細胞をあらかじめ移入していた場合にも,酪酸化デンプン摂取によりOT-IIマウス由来のナイーブT細胞から分化したpTreg細胞数が増加していたことから,酪酸は大腸粘膜局所においてナイーブT細胞からpTreg細胞への分化を誘導することが明らかとなった17).
米国のWendy Garrettらの研究グループは,高濃度の短鎖脂肪酸を飲水に混合してSPFマウスや無菌マウスに3週間自由摂取させることで,大腸粘膜において胸腺由来のTreg細胞(thymus-derived Treg cell:tTreg細胞)が増加することを報告している48).興味深いことにこの場合には,プロピオン酸や酢酸には強いtTreg細胞誘導作用が認められたものの,酪酸の効果は弱かった.この結果については,短鎖脂肪酸を飲水摂取するとそのほとんどが大腸に到達する以前に小腸で吸収されてしまうため,腸内細菌による生理的な短鎖脂肪酸産生の場である大腸局所における機能は検討できず,さらに非生理的高濃度の短鎖脂肪酸が血中に流入してしまうことによる影響である可能性に留意する必要がある.実際に上記の報告では,酢酸やプロピオン酸は,tTreg細胞が発現しているGタンパク質共役型受容体43(G-protein-coupled receptor 43:GPR43)に作用することで,ナイーブT細胞からの分化ではなく,大腸への遊走を促進すると考察しており,これは全身性の作用といえる.プロピオン酸を飲水摂取したマウスのtTreg細胞は,大腸ホーミング因子であるGPR15の遺伝子発現を高めることでその機能を獲得していることも示唆されている48, 49).
筆者らは前述のin vitro実験の結果から,酪酸(および活性は弱いながらもプロピオン酸)は,ナイーブT細胞に直接作用することでTreg細胞への分化を促進することを示した.一方で米国のRudenskyらのグループは,酪酸によるナイーブT細胞への直接的な作用に加えて,酪酸が樹状細胞に作用することで間接的にTreg細胞への分化を促進する可能性も報告している50).Flt3リガンドによる刺激で誘導した樹状細胞を酪酸で前処理し,新鮮な培地に変えてナイーブT細胞と共培養すると,Treg細胞への分化が促進された50).この場合,酪酸が樹状細胞における炎症性サイトカインやRelBの発現を抑制することで,Treg細胞への分化を促進していることが示唆されている.
では,酪酸は大腸粘膜においてどのようにナイーブT細胞からpTreg細胞への分化を誘導するのだろうか? これまでの報告から,酪酸はヒストン脱アセチル化酵素(histone deacetylase:HDAC)阻害剤として機能することが知られている51, 52).酪酸と比較して活性は弱いながらも,プロピオン酸にもHDAC阻害活性は認められているが,一方で酢酸にはその活性がほとんどない.このように各短鎖脂肪酸のHDAC阻害活性(酪酸>プロピオン酸≫酢酸)とTreg細胞誘導活性には正の相関があったことから,酪酸はナイーブT細胞のヒストンアセチル化状態を変化させることで,Treg細胞への分化誘導を促進する可能性が示唆された.HDACは,ヒストンテイルにおけるリジン残基の脱アセチル化を促進し,転写を負に調節するコリプレッサーであるため,酪酸がHDACの阻害作用を介してヒストンアセチル化に影響を与えていると考えられた.そこで,クロマチン免疫沈降-シークエンス(chromatin immunoprecipitation-sequence:ChIP-seq)法によりヒストンアセチル化についてゲノムワイドに解析した.その結果,in vitroでのナイーブT細胞のTreg細胞分化誘導条件下で酪酸を添加しておくと,Treg細胞のマスター転写因子であるFoxp3遺伝子上流のプロモーター領域およびイントロンに存在するCNS(conserved non-coding sequence)3エンハンサー領域におけるヒストンH3のアセチル化が亢進することが明らかとなった17).トランスクリプトーム解析結果から,このようなエピジェネティックな変化は,Foxp3遺伝子の発現誘導に先立って誘導されることが明らかとなった17).一方,興味深いことに,エフェクターT細胞のマスター転写因子であるTbx21(T-bet)やGata3, Rorc(ROR γt)遺伝子プロモーターのアセチル化は亢進せず,TH1やTH2, TH17などのエフェクターT細胞への分化には影響しなかったことから,酪酸はナイーブT細胞において選択的にFoxp3発現を誘導していると結論づけられる.一方Rudenskyらは,酪酸によるFoxp3タンパク質のアセチル化の亢進の可能性も示唆している50).
続いて,酪酸で誘導されたTreg細胞が実際に生体内で機能的であるか否かの検証を試みた.SPFマウスに酪酸化デンプン含有飼料を与えた際に増加するpTreg細胞は,抑制性サイトカインであるIL-10を産生することが明らかとなった17).次に,CD4+CD45RBhiナイーブT細胞をRag1欠損マウスに移入することで,IBDの実験的モデルとして知られているヘルパーT細胞介在性の慢性大腸炎モデルマウスを作製し,酪酸化デンプン含有飼料または非含有の対照飼料を与えた.その結果,酪酸化デンプン含有飼料摂取群では大腸粘膜におけるpTreg細胞の割合が増加し,腸炎発症時に観察される体重減少,大腸粘膜への炎症性細胞浸潤,および腸管壁肥厚の軽減が認められた17).次に,Foxp3遺伝子の転写制御下にhuman CD2(hCD2)を発現するレポーターマウス由来のナイーブT細胞を用いて同様の大腸炎モデルを作出した後,抗hCD2抗体投与によりTreg細胞を除去したところ,酪酸化デンプン摂取による抗炎症作用が完全に消失した17).この結果から,酪酸による炎症抑制効果はTreg細胞の分化誘導を介することが確認された.以上の知見より,クロストリジウム目細菌群などによる腸内発酵を介して産生される酪酸が,Foxp3遺伝子のヒストンアセチル化を促進することで機能的なTreg細胞を誘導し,腸管の免疫恒常性の維持に大きく寄与していることが明らかとなった(図2).
2)腸内細菌が産生する代謝物質は多様な生体修飾機構を有する
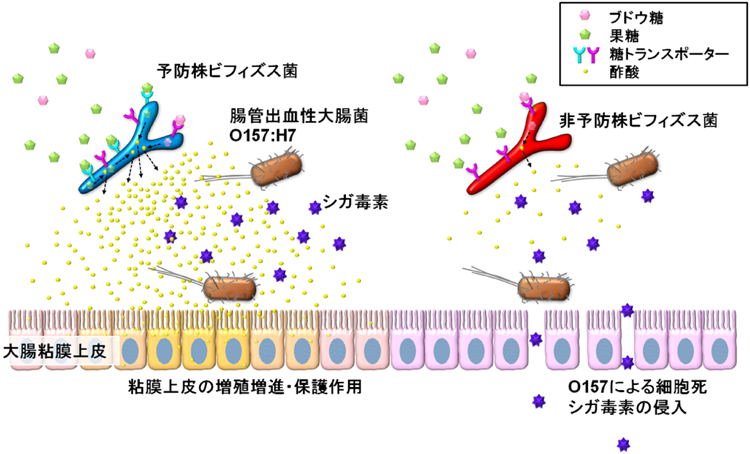
上述の酪酸以外にも,腸内細菌が腸管内で産生する短鎖脂肪酸や,乳酸などの有機酸には生体修飾因子としての機能があることが報告されている.たとえば腸内細菌叢由来の酢酸は腸から血中に移行し,白血球の一種であり炎症反応の中心的役割を担う好中球が発現しているGPR43を介してアポトーシスを促し,大腸炎モデルマウスにおいて大腸炎を抑制することが報告されている53).筆者らも,腸管出血性大腸菌O157:H7感染マウスモデルにおいて,腸内細菌の一種でありプロバイオティクスとしても利用されているビフィズス菌によるO157腸管感染症予防機構について解析を行ったところ,ビフィズス菌が腸管内で産生する酢酸が,宿主腸管上皮細胞のバリア機能を高めることで,O157による腸管感染症を予防できることを明らかにした(図3)54).
短鎖脂肪酸と2型糖尿病との関係も示唆されており,不溶性食物繊維が腸内細菌叢により代謝され産生された短鎖脂肪酸が,脂肪細胞のペルオキシソーム増殖因子活性化受容体γ(peroxisome proliferator-activated receptor γ:PPARγ)経路を活性化することで,インスリン抵抗性を改善し,2型糖尿病発症率を低下させることも報告されている55).また脂肪細胞が発現しているGPR43に腸内細菌叢由来短鎖脂肪酸が作用し,脂肪細胞のインスリンシグナルを抑制することで脂肪蓄積の抑制,および他の臓器における脂質や糖質の代謝を促すことで,生体内の代謝恒常性を維持していることも報告されている56).膵臓のβ細胞が発現するGPR43にも腸内細菌叢由来の酢酸が作用し,インスリン分泌を促進することが報告されているが57),高脂肪食摂取により作製した2型糖尿病モデルマウスの膵臓β細胞においては,腸内細菌叢由来の酢酸がβ細胞のGPR41やGPR43を介して,逆にインスリン分泌を抑制し,結果として耐糖能を悪化させるといった報告もあることから58),膵臓のβ細胞におけるGPR41やGPR43を介した腸内細菌叢由来酢酸の2型糖尿病への関与は議論の余地が残されている.腸管局所での機能としては,マウスにおいて短鎖脂肪酸が結腸上皮に存在する消化管L細胞が発現するGPR41やGPR43を介して,L細胞から消化管ホルモンの一つであるグルカゴン様ペプチド1(Glucagon-like peptide-1:GLP-1)の分泌を促し,膵臓からのインスリン分泌や食欲抑制作用を促すことが報告されている59).短鎖脂肪酸の中でも特にプロピオン酸がヒト腸管内で作用し,peptide YY(PYY)やGLP-1などの消化管ホルモンの血中濃度の増加に寄与することが報告されており60),腸内細菌叢由来短鎖脂肪酸が,抗肥満や糖尿病予防の新たな標的になる可能性がある.
他にも筆者らは,腸内細菌が産生する有機酸の一つである乳酸が,宿主の絶食後の再摂食時に生じる大腸上皮細胞の過増殖を促す鍵因子であることを明らかにしており,大腸がん誘発モデルにおいて,絶食-再摂食時の乳酸注腸による大腸上皮の過増殖が,前がん病変の増加につながることも報告している61).このように腸内細菌が産生する短鎖脂肪酸や有機酸には多様な生体修飾因子としての機能があることが明らかになりつつあるが,これらは腸内細菌叢機能の一側面にすぎないと考えられることから,メタボロゲノミクスによる腸内細菌叢機能の全容理解に向けてさらなる研究進展が望まれる.
引用文献References
1) Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K.S., Manichanh, C., Nielsen, T., Pons, N., Levenez, F., Yamada, T., Mende, D.R., Li, J., Xu, J., Li, S., Li, D., Cao, J., Wang, B., Liang, H., Zheng, H., Xie, Y., Tap, J., Lepage, P., Bertalan, M., Batto, J.M., Hansen, T., Le Paslier, D., Linneberg, A., Nielsen, H.B., Pelletier, E., Renault, P., Sicheritz-Ponten, T., Turner, K., Zhu, H., Yu, C., Li, S., Jian, M., Zhou, Y., Li, Y., Zhang, X., Li, S., Qin, N., Yang, H., Wang, J., Brunak, S., Dore, J., Guarner, F., Kristiansen, K., Pedersen, O., Parkhill, J., Weissenbach, J., Meta, H.I.T.C., Bork, P., Ehrlich, S.D., & Wang, J. (2010) Nature, 464, 59–65.
2) Bianconi, E., Piovesan, A., Facchin, F., Beraudi, A., Casadei, R., Frabetti, F., Vitale, L., Pelleri, M.C., Tassani, S., Piva, F., Perez-Amodio, S., Strippoli, P., & Canaider, S. (2013) Ann. Hum. Biol., 40, 463–471.
3) Fukuda, S. & Ohno, H. (2014) Semin. Immunopathol., 36, 103–114.
4) Fukuda, S., Nakanishi, Y., Chikayama, E., Ohno, H., Hino, T., & Kikuchi, J. (2009) PLoS ONE, 4, e4893.
5) Nakanishi, Y., Fukuda, S., Chikayama, E., Kimura, Y., Ohno, H., & Kikuchi, J. (2011) J. Proteome Res., 10, 824–836.
6) Buscher, J.M., Czernik, D., Ewald, J.C., Sauer, U., & Zamboni, N. (2009) Anal. Chem., 81, 2135–2143.
7) Soga, T., Ohashi, Y., Ueno, Y., Naraoka, H., Tomita, M., & Nishioka, T. (2003) J. Proteome Res., 2, 488–494.
8) Ohashi, Y., Hirayama, A., Ishikawa, T., Nakamura, S., Shimizu, K., Ueno, Y., Tomita, M., & Soga, T. (2008) Mol. Biosyst., 4, 135–147.
9) Steinhauser, M.L., Bailey, A.P., Senyo, S.E., Guillermier, C., Perlstein, T.S., Gould, A.P., Lee, R.T., & Lechene, C.P. (2012) Nature, 481, 516–519.
10) Taguchi, K., Fukusaki, E., & Bamba, T. (2014) Bioanalysis, 6, 1679–1689.
11) Barrett, J.C., Hansoul, S., Nicolae, D.L., Cho, J.H., Duerr, R.H., Rioux, J.D., Brant, S.R., Silverberg, M.S., Taylor, K.D., Barmada, M.M., Bitton, A., Dassopoulos, T., Datta, L.W., Green, T., Griffiths, A.M., Kistner, E.O., Murtha, M.T., Regueiro, M.D., Rotter, J.I., Schumm, L.P., Steinhart, A.H., Targan, S.R., Xavier, R.J., Libioulle, C., Sandor, C., Lathrop, M., Belaiche, J., Dewit, O., Gut, I., Heath, S., Laukens, D., Mni, M., Rutgeerts, P., Van Gossum, A., Zelenika, D., Franchimont, D., Hugot, J.P., de Vos, M., Vermeire, S., Louis, E., Cardon, L.R., Anderson, C.A., Drummond, H., Nimmo, E., Ahmad, T., Prescott, N.J., Onnie, C.M., Fisher, S.A., Marchini, J., Ghori, J., Bumpstead, S., Gwilliam, R., Tremelling, M., Deloukas, P., Mansfield, J., Jewell, D., Satsangi, J., Mathew, C.G., Parkes, M., Georges, M., & Daly, M.J.; NIDDK IBD Genetics Consortium; Belgian-French IBD Consortium; Wellcome Trust Case Control Consortium (2008) Nat. Genet., 40, 955–962.
12) Frank, D.N., St Amand, A.L., Feldman, R.A., Boedeker, E.C., Harpaz, N., & Pace, N.R. (2007) Proc. Natl. Acad. Sci. USA, 104, 13780–13785.
13) Sokol, H., Pigneur, B., Watterlot, L., Lakhdari, O., Bermudez-Humaran, L.G., Gratadoux, J.J., Blugeon, S., Bridonneau, C., Furet, J.P., Corthier, G., Grangette, C., Vasquez, N., Pochart, P., Trugnan, G., Thomas, G., Blottiere, H.M., Dore, J., Marteau, P., Seksik, P., & Langella, P. (2008) Proc. Natl. Acad. Sci. USA, 105, 16731–16736.
14) Norman, J.M., Handley, S.A., Baldridge, M.T., Droit, L., Liu, C.Y., Keller, B.C., Kambal, A., Monaco, C.L., Zhao, G., Fleshner, P., Stappenbeck, T.S., McGovern, D.P., Keshavarzian, A., Mutlu, E.A., Sauk, J., Gevers, D., Xavier, R.J., Wang, D., Parkes, M., & Virgin, H.W. (2015) Cell, 160, 447–460.
15) Atarashi, K., Tanoue, T., Shima, T., Imaoka, A., Kuwahara, T., Momose, Y., Cheng, G., Yamasaki, S., Saito, T., Ohba, Y., Taniguchi, T., Takeda, K., Hori, S., Ivanov, I.I., Umesaki, Y., Itoh, K., & Honda, K. (2011) Science, 331, 337–341.
16) Atarashi, K., Tanoue, T., Oshima, K., Suda, W., Nagano, Y., Nishikawa, H., Fukuda, S., Saito, T., Narushima, S., Hase, K., Kim, S., Fritz, J.V., Wilmes, P., Ueha, S., Matsushima, K., Ohno, H., Olle, B., Sakaguchi, S., Taniguchi, T., Morita, H., Hattori, M., & Honda, K. (2013) Nature, 500, 232–236.
17) Furusawa, Y., Obata, Y., Fukuda, S., Endo, T.A., Nakato, G., Takahashi, D., Nakanishi, Y., Uetake, C., Kato, K., Kato, T., Takahashi, M., Fukuda, N.N., Murakami, S., Miyauchi, E., Hino, S., Atarashi, K., Onawa, S., Fujimura, Y., Lockett, T., Clarke, J.M., Topping, D.L., Tomita, M., Hori, S., Ohara, O., Morita, T., Koseki, H., Kikuchi, J., Honda, K., Hase, K., & Ohno, H. (2013) Nature, 504, 446–450.
18) Arthur, J.C., Perez-Chanona, E., Muhlbauer, M., Tomkovich, S., Uronis, J.M., Fan, T.J., Campbell, B.J., Abujamel, T., Dogan, B., Rogers, A.B., Rhodes, J.M., Stintzi, A., Simpson, K.W., Hansen, J.J., Keku, T.O., Fodor, A.A., & Jobin, C. (2012) Science, 338, 120–123.
19) Yoshimoto, S., Loo, T.M., Atarashi, K., Kanda, H., Sato, S., Oyadomari, S., Iwakura, Y., Oshima, K., Morita, H., Hattori, M., Honda, K., Ishikawa, Y., Hara, E., & Ohtani, N. (2013) Nature, 499, 97–101.
20) Belcheva, A., Irrazabal, T., Robertson, S.J., Streutker, C., Maughan, H., Rubino, S., Moriyama, E.H., Copeland, J.K., Kumar, S., Green, B., Geddes, K., Pezo, R.C., Navarre, W.W., Milosevic, M., Wilson, B.C., Girardin, S.E., Wolever, T.M., Edelmann, W., Guttman, D.S., Philpott, D.J., & Martin, A. (2014) Cell, 158, 288–299.
21) Schulz, M.D., Atay, C., Heringer, J., Romrig, F.K., Schwitalla, S., Aydin, B., Ziegler, P.K., Varga, J., Reindl, W., Pommerenke, C., Salinas-Riester, G., Bock, A., Alpert, C., Blaut, M., Polson, S.C., Brandl, L., Kirchner, T., Greten, F.R., Polson, S.W., & Arkan, M.C. (2014) Nature, 514, 508–512.
22) Ghoshal, U.C., Park, H., & Gwee, K.A. (2010) J. Gastroenterol. Hepatol., 25, 244–251.
23) Yano, J.M., Yu, K., Donaldson, G.P., Shastri, G.G., Ann, P., Ma, L., Nagler, C.R., Ismagilov, R.F., Mazmanian, S.K., & Hsiao, E.Y. (2015) Cell, 161, 264–276.
24) Aw, W. & Fukuda, S. (2015) Semin. Immunopathol., 37, 5–16.
25) Turnbaugh, P.J., Ley, R.E., Mahowald, M.A., Magrini, V., Mardis, E.R., & Gordon, J.I. (2006) Nature, 444, 1027–1031.
26) Membrez, M., Blancher, F., Jaquet, M., Bibiloni, R., Cani, P.D., Burcelin, R.G., Corthesy, I., Mace, K., & Chou, C.J. (2008) FASEB J., 22, 2416–2426.
27) Ridaura, V.K., Faith, J.J., Rey, F.E., Cheng, J., Duncan, A.E., Kau, A.L., Griffin, N.W., Lombard, V., Henrissat, B., Bain, J.R., Muehlbauer, M.J., Ilkayeva, O., Semenkovich, C.F., Funai, K., Hayashi, D.K., Lyle, B.J., Martini, M.C., Ursell, L.K., Clemente, J.C., Van Treuren, W., Walters, W.A., Knight, R., Newgard, C.B., Heath, A.C., & Gordon, J.I. (2013) Science, 341, 1241214.
28) Everard, A., Lazarevic, V., Derrien, M., Girard, M., Muccioli, G.G., Neyrinck, A.M., Possemiers, S., Van Holle, A., Francois, P., de Vos, W.M., Delzenne, N.M., Schrenzel, J., & Cani, P.D. (2011) Diabetes, 60, 2775–2786.
29) Everard, A., Belzer, C., Geurts, L., Ouwerkerk, J.P., Druart, C., Bindels, L.B., Guiot, Y., Derrien, M., Muccioli, G.G., Delzenne, N.M., de Vos, W.M., & Cani, P.D. (2013) Proc. Natl. Acad. Sci. USA, 110, 9066–9071.
30) Goodrich, J.K., Waters, J.L., Poole, A.C., Sutter, J.L., Koren, O., Blekhman, R., Beaumont, M., Van Treuren, W., Knight, R., Bell, J.T., Spector, T.D., Clark, A.G., & Ley, R.E. (2014) Cell, 159, 789–799.
31) Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., Liang, S., Zhang, W., Guan, Y., Shen, D., Peng, Y., Zhang, D., Jie, Z., Wu, W., Qin, Y., Xue, W., Li, J., Han, L., Lu, D., Wu, P., Dai, Y., Sun, X., Li, Z., Tang, A., Zhong, S., Li, X., Chen, W., Xu, R., Wang, M., Feng, Q., Gong, M., Yu, J., Zhang, Y., Zhang, M., Hansen, T., Sanchez, G., Raes, J., Falony, G., Okuda, S., Almeida, M., LeChatelier, E., Renault, P., Pons, N., Batto, J.M., Zhang, Z., Chen, H., Yang, R., Zheng, W., Li, S., Yang, H., Wang, J., Ehrlich, S.D., Nielsen, R., Pedersen, O., Kristiansen, K., & Wang, J. (2012) Nature, 490, 55–60.
32) Karlsson, F.H., Tremaroli, V., Nookaew, I., Bergstrom, G., Behre, C.J., Fagerberg, B., Nielsen, J., & Backhed, F. (2013) Nature, 498, 99–103.
33) Koeth, R.A., Wang, Z., Levison, B.S., Buffa, J.A., Org, E., Sheehy, B.T., Britt, E.B., Fu, X., Wu, Y., Li, L., Smith, J.D., DiDonato, J.A., Chen, J., Li, H., Wu, G.D., Lewis, J.D., Warrier, M., Brown, J.M., Krauss, R.M., Tang, W.H., Bushman, F.D., Lusis, A.J., & Hazen, S.L. (2013) Nat. Med., 19, 576–585.
34) Tang, W.H., Wang, Z., Levison, B.S., Koeth, R.A., Britt, E.B., Fu, X., Wu, Y., & Hazen, S.L. (2013) N. Engl. J. Med., 368, 1575–1584.
35) Suez, J., Korem, T., Zeevi, D., Zilberman-Schapira, G., Thaiss, C.A., Maza, O., Israeli, D., Zmora, N., Gilad, S., Weinberger, A., Kuperman, Y., Harmelin, A., Kolodkin-Gal, I., Shapiro, H., Halpern, Z., Segal, E., & Elinav, E. (2014) Nature, 514, 181–186.
36) Chassaing, B., Koren, O., Goodrich, J.K., Poole, A.C., Srinivasan, S., Ley, R.E., & Gewirtz, A.T. (2015) Nature, 519, 92–96.
37) Hsiao, E.Y., McBride, S.W., Hsien, S., Sharon, G., Hyde, E.R., McCue, T., Codelli, J.A., Chow, J., Reisman, S.E., Petrosino, J.F., Patterson, P.H., & Mazmanian, S.K. (2013) Cell, 155, 1451–1463.
38) Aronov, P.A., Luo, F.J., Plummer, N.S., Quan, Z., Holmes, S., Hostetter, T.H., & Meyer, T.W. (2011) J. Am. Soc. Nephrol., 22, 1769–1776.
39) Mishima, E., Fukuda, S., Shima, H., Hirayama, A., Akiyama, Y., Takeuchi, Y., Fukuda, N.N., Suzuki, T., Suzuki, C., Yuri, A., Kikuchi, K., Tomioka, Y., Ito, S., Soga, T., & Abe, T. (2015) J. Am. Soc. Nephrol., 26, 1787–1794.
40) Yokote, H., Miyake, S., Croxford, J.L., Oki, S., Mizusawa, H., & Yamamura, T. (2008) Am. J. Pathol., 173, 1714–1723.
41) Miyake, S., Kim, S., Suda, W., Oshima, K., Nakamura, M., Matsuoka, T., Chihara, N., Tomita, A., Sato, W., Kim, S.W., Morita, H., Hattori, M., & Yamamura, T. (2015) PLoS ONE, 10, e0137429.
42) Cahenzli, J., Koller, Y., Wyss, M., Geuking, M.B., & McCoy, K.D. (2013) Cell Host Microbe, 14, 559–570.
43) Umesaki, Y., Setoyama, H., Matsumoto, S., Imaoka, A., & Itoh, K. (1999) Infect. Immun., 67, 3504–3511.
44) Ivanov, I.I., Atarashi, K., Manel, N., Brodie, E.L., Shima, T., Karaoz, U., Wei, D., Goldfarb, K.C., Santee, C.A., Lynch, S.V., Tanoue, T., Imaoka, A., Itoh, K., Takeda, K., Umesaki, Y., Honda, K., & Littman, D.R. (2009) Cell, 139, 485–498.
45) Goto, Y., Panea, C., Nakato, G., Cebula, A., Lee, C., Diez, M.G., Laufer, T.M., Ignatowicz, L., & Ivanov, I.I. (2014) Immunity, 40, 594–607.
46) Yang, Y., Torchinsky, M.B., Gobert, M., Xiong, H., Xu, M., Linehan, J.L., Alonzo, F., Ng, C., Chen, A., Lin, X., Sczesnak, A., Liao, J.J., Torres, V.J., Jenkins, M.K., Lafaille, J.J., & Littman, D.R. (2014) Nature, 510, 152–156.
47) Schnupf, P., Gaboriau-Routhiau, V., Gros, M., Friedman, R., Moya-Nilges, M., Nigro, G., Cerf-Bensussan, N., & Sansonetti, P.J. (2015) Nature, 520, 99–103.
48) Smith, P.M., Howitt, M.R., Panikov, N., Michaud, M., Gallini, C.A., Bohlooly, Y.M., Glickman, J.N., & Garrett, W.S. (2013) Science, 341, 569–573.
49) Kim, S.V., Xiang, W.V., Kwak, C., Yang, Y., Lin, X.W., Ota, M., Sarpel, U., Rifkin, D.B., Xu, R., & Littman, D.R. (2013) Science, 340, 1456–1459.
50) Arpaia, N., Campbell, C., Fan, X., Dikiy, S., van der Veeken, J., deRoos, P., Liu, H., Cross, J.R., Pfeffer, K., Coffer, P.J., & Rudensky, A.Y. (2013) Nature, 504, 451–455.
51) Candido, E.P., Reeves, R., & Davie, J.R. (1978) Cell, 14, 105–113.
52) Davie, J.R. (2003) J. Nutr., 133(Suppl), 2485S–2493S.
53) Maslowski, K.M., Vieira, A.T., Ng, A., Kranich, J., Sierro, F., Yu, D., Schilter, H.C., Rolph, M.S., Mackay, F., Artis, D., Xavier, R.J., Teixeira, M.M., & Mackay, C.R. (2009) Nature, 461, 1282–1286.
54) Fukuda, S., Toh, H., Hase, K., Oshima, K., Nakanishi, Y., Yoshimura, K., Tobe, T., Clarke, J.M., Topping, D.L., Suzuki, T., Taylor, T.D., Itoh, K., Kikuchi, J., Morita, H., Hattori, M., & Ohno, H. (2011) Nature, 469, 543–547.
55) Robertson, M.D., Wright, J.W., Loizon, E., Debard, C., Vidal, H., Shojaee-Moradie, F., Russell-Jones, D., & Umpleby, A.M. (2012) J. Clin. Endocrinol. Metab., 97, 3326–3332.
56) Kimura, I., Ozawa, K., Inoue, D., Imamura, T., Kimura, K., Maeda, T., Terasawa, K., Kashihara, D., Hirano, K., Tani, T., Takahashi, T., Miyauchi, S., Shioi, G., Inoue, H., & Tsujimoto, G. (2013) Nat. Commun., 4, 1829.
57) McNelis, J.C., Lee, Y.S., Mayoral, R., van der Kant, R., Johnson, A.M., Wollam, J., & Olefsky, J.M. (2015) Diabetes, 64, 3203–3217.
58) Tang, C., Ahmed, K., Gille, A., Lu, S., Grone, H.J., Tunaru, S., & Offermanns, S. (2015) Nat. Med., 21, 173–177.
59) Tolhurst, G., Heffron, H., Lam, Y.S., Parker, H.E., Habib, A.M., Diakogiannaki, E., Cameron, J., Grosse, J., Reimann, F., & Gribble, F.M. (2012) Diabetes, 61, 364–371.
60) Chambers, E.S., Viardot, A., Psichas, A., Morrison, D.J., Murphy, K.G., Zac-Varghese, S.E., MacDougall, K., Preston, T., Tedford, C., Finlayson, G.S., Blundell, J.E., Bell, J.D., Thomas, E.L., Mt-Isa, S., Ashby, D., Gibson, G.R., Kolida, S., Dhillo, W.S., Bloom, S.R., Morley, W., Clegg, S., & Frost, G. (2015) Gut, 64, 1744–1754.
61) Okada, T., Fukuda, S., Hase, K., Nishiumi, S., Izumi, Y., Yoshida, M., Hagiwara, T., Kawashima, R., Yamazaki, M., Oshio, T., Otsubo, T., Inagaki-Ohara, K., Kakimoto, K., Higuchi, K., Kawamura, Y.I., Ohno, H., & Dohi, T. (2013) Nat. Commun., 4, 1654.