Arfは,主に細胞内小胞輸送やアクチン細胞骨格の再構成を制御する低分子量Gタンパク質である.ArfファミリーにはArf1~6の六つのアイソフォームが存在し,それらはアミノ酸配列の相同性に基づき,Arf1~3が属するクラスI,Arf4および5が属するクラスII,Arf6が属するクラスIIIの三つのクラスに分類されている1).クラスIとクラスIIに属するArfアイソフォームは,主にゴルジ体や小胞体に局在し,唯一クラスIIIに分類されるArf6は,エンドソームや細胞膜に局在している2).Arf6とその他のArfアイソフォームの細胞内局在の違いは,Arf6がその他のArf分子と機能的に一線を画する要因となっている.すなわち,クラスIとIIに属するArfは,主に小胞体やゴルジ体においてコートタンパク質複合体の形成を誘導することにより小胞体–ゴルジ体間やゴルジ嚢間の小胞輸送を制御している.一方,クラスIIIのArf6は,細胞膜やエンドソームにおいて,細胞膜–エンドソーム間の小胞輸送や,アクチン細胞骨格の再構成を介した細胞膜ダイナミクスに関与していることが知られている2).Arf6が制御する細胞機能は,生命現象の根幹をなす重要な機能であり,本稿の「3.細胞レベルでのArf6の生理機能」の項で述べるように,Arf6は実に多様な細胞機能に関与している.さらに近年,我々が作製したArf6遺伝子ノックアウト(KO)マウスを用いた解析やArf6シグナル伝達系阻害剤のマウスへの投与実験によって,個体レベルでのArf6の生理機能や疾患との関わりも徐々に明らかになりつつある.本稿では,多彩な生理機能を有するArf6のシグナル伝達機構および細胞レベルでのArf6の生理機能を概説し,近年明らかとなってきた個体レベルでのArf6の役割について,我々の最新の知見を含めて紹介する.
1)Arf6の活性調節機構
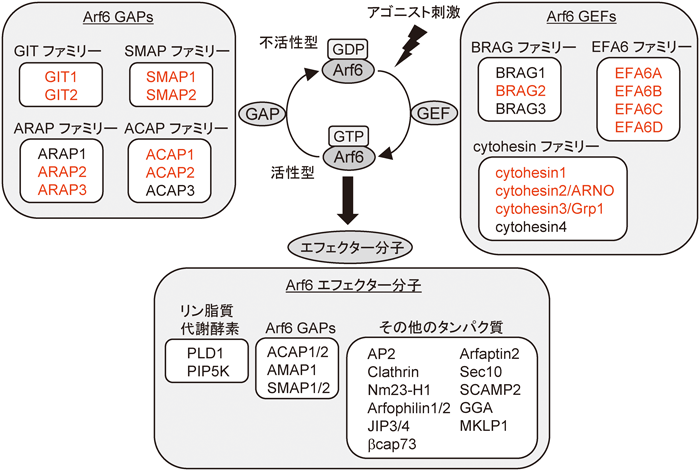
定常状態の細胞では,Arf6はGDPを結合した不活性型で存在しているが,細胞が成長因子やホルモンなどの刺激を受容すると,Arf6に結合しているGDPが解離し,その部位にGTPが新たに結合することにより活性型となる(図1).このグアニンヌクレオチドの交換は,アゴニスト刺激に応答して活性化されるグアニンヌクレオチド交換因子(guanine nucleotide exchange factor:GEF)の作用により引き起こされる.活性化されたArf6は,さまざまな下流分子(エフェクター分子)と相互作用することによってシグナルを下流に伝達する.その後,Arf6は自身の有するGTPase活性により結合したGTPをGDPに加水分解して,再び不活性化状態へ戻るため,エフェクター分子がArf6から解離して上流からのシグナル伝達は遮断される.通常,低分子量Gタンパク質が有するGTPase活性は非常に低いため,Arf6が結合しているGTPをGDPに加水分解するためには,GTPase活性化因子(GTPase activating protein:GAP)の補助が必要である.
Arf6のGDP-GTP交換反応を触媒するGEFとしては,これまでにBRAGファミリー,EFA6ファミリー,およびcytohesinファミリーの三つのファミリーが同定されている3)(図1).BRAGファミリーはBRAG1~3の三つの分子が存在し,そのうちBRAG2がArf6に対するGEF活性を有している.EFA6ファミリーはEFA6A~Dの四つのアイソフォームで構成されており,それらのすべてがArf6のGEFとして機能する.cytohesinファミリーとして同定されているcytohesin1~4の四つの分子のうち,cytohesin1~3がArf6 GEFとして機能する.
Arf6を不活性化するタンパク質群であるGAPにおいても複数の分子が同定されている(図1).in vitroのGAP活性測定実験において,GIT1, GIT2, SMAP1, SMAP2, ARAP2, ARAP3, ACAP1, ACAP2などの多数の分子がArf6のGAPとして機能することが報告されている4–6).このように多数のArf6 GEFおよびGAPが存在する理由は,それぞれのGEFおよびGAPが特定の細胞内部位で特定のシグナル伝達経路においてArf6を活性化・不活性化することによってArf6を介したシグナル伝達系に多様性を持たせるためであると推測できる.
2)Arf6エフェクター分子
図1に示すように,これまでにArf6が制御する多種多様なエフェクター分子が同定されている.Arf6は,これらのエフェクター分子の細胞内局在を制御するか,あるいは活性状態を変化させることで下流にシグナルを伝達し,特定の細胞機能の発揮に寄与している.これらのArf6エフェクター分子は,主にリン脂質代謝酵素,Arf6 GAP,その他のタンパク質の三つに分類することができる.
Arf6は,ホスファチジン酸(phosphatidic acid:PA)を産生するホスホリパーゼD1(phospholipase D1:PLD1)や,ホスファチジルイノシトール4,5-二リン酸(phosphatidylinositol 4,5-bisphosphate:PI(4,5)P2)を産生するホスファチジルイノシトール4-リン酸5-キナーゼ(phosphatidylinositol 4-phosphate 5-kinase:PIP5K)などのリン脂質代謝酵素と直接結合してそれらを活性化する7).これらのリン脂質代謝酵素の代謝産物であるPAやPI(4,5)P2は,これらの脂質に結合するさまざまなタンパク質・酵素を制御する脂質性セカンドメッセンジャーとして機能しており,このこともArf6が多様な生理機能を担う理由の一端であると考えられる.
GAPは通常,Arf6を不活性化するタンパク質群であるが,いくつかのGAPについてはArf6のエフェクター分子としての機能を有していることが明らかとなっている.それらのGAPは,それぞれ特有の分子と相互作用することにより,活性型Arf6が存在する部位にGAP結合タンパク質をリクルートする機能を有していると考えられている.このようなGAPとしては,ACAP1, SMAP1/2, AMAP1などが知られている5, 6, 8, 9).それぞれのGAPは,そのドメイン構造が大きく異なり,それぞれ特異的な結合タンパク質を有しているため10),上記以外のGAPに関してもその結合タンパク質を介してArf6のエフェクター分子として機能する可能性が十分に考えられる.
上記の分子以外に,図1にあげるようにArf6は実にさまざまなタンパク質と結合することが知られている.それらはArf6の関与する細胞機能に直結したエフェクター分子である場合が多く,その細胞機能としては細胞内小胞輸送とアクチン細胞骨格の再構成があげられる.このように多様なエフェクター分子が存在することによって,Arf6がさまざまな生理機能を発揮することが可能となっている.
Arf6は多岐にわたるエフェクター分子と相互作用することによって,さまざまな細胞機能を制御することが知られている.Arf6が制御する細胞機能としては,細胞内小胞輸送とアクチン細胞骨格の再構成を伴う細胞膜ダイナミクスの二つの現象に大別される.
1)細胞内小胞輸送
Arf6はこれまでに,さまざまな細胞膜タンパク質のクラスリン依存的あるいは非依存的エンドサイトーシスに関与することが報告されている2).クラスリン依存的エンドサイトーシスに関しては,β2アドレナリン受容体やトランスフェリン受容体,細胞接着因子のE-カドヘリンなどのエンドサイトーシスがArf6により制御されている11–13).この現象において,Arf6の果たす役割は多面的であると考えられている.Arf6のエフェクター分子であるPIP5Kにより細胞膜で産生されたPI(4,5)P2は,AP2やAP180,ダイナミンなど,複数のエンドサイトーシス関連タンパク質と結合して,これらのタンパク質をエンドサイトーシス部位へリクルートすることによりエンドサイトーシスを誘起する14).トランスフェリン受容体のエンドサイトーシスに関して,Arf6 GAPであるSMAP1がArf6のエフェクター分子として機能し,クラスリンの細胞膜へのリクルートに関与している5).さらに,Arf6自身がAP2やクラスリンと結合することが示されており15),これらの下流分子を介してArf6はクラスリン依存性のエンドサイトーシスを複合的に制御している.クラスリン非依存性のエンドサイトーシスにおいては,抗原提示に重要な役割を果たすMHCクラスIや細胞外マトリックスに対する接着分子のインテグリンなどの細胞膜タンパク質のエンドサイトーシスがArf6の制御を受けるが2),現在のところその詳細な分子メカニズムは不明である.
エンドサイトーシスされた膜タンパク質の一部は,その後エンドソームから細胞膜へ再びリサイクリングされて再利用される.Arf6はこれまでに,細胞外マトリックスに対する接着分子β1インテグリン,グルコース輸送体4 (glucose transporter 4:Glut4),細胞膜貫通型ヘパラン硫酸プロテオグリカンであるシンデカンなどのタンパク質のリサイクリングを制御することが報告されている16–18).この現象においては,Arf6 GAPの一つであるACAP1がArf6のエフェクター分子として重要な機能を果たす17).ACAP1は,Glut4やβ1インテグリンなどの積荷分子とエンドソームで結合する.さらに,ACAP1はクラスリンとも結合することが示されており,Arf6-ACAP1複合体は,エンドソームにおいて積荷タンパク質を含んだクラスリン被服小胞の形成に寄与していると考えられる.また,Arf6は細胞膜への小胞輸送を制御するexocyst複合体のサブユニットの一つであるSec10と結合することも知られており19),膜タンパク質のリサイクリングにおいてもArf6が機能しているステップは複数存在すると考えられる.
Arf6は,細胞内小胞輸送を介してファゴサイトーシスや細胞分裂においても機能している20, 21).マクロファージにおけるファゴソームの形成過程では,アクチン細胞骨格の再構成を介した細胞膜の伸長と,細胞内膜系から細胞膜への膜輸送による膜成分の供給が必要である.この現象において,Arf6はファゴサイトーシス部位へのエンドソームからの膜供給に貢献していることが示されている20).また,細胞分裂において一つの細胞が二つの娘細胞に分裂する際に,細胞内膜系から細胞膜への膜供給が必要であるが,Arf6は細胞分裂時に形成されるミッドボディに局在し,エンドソームから細胞膜への膜供給を担うというモデルが提唱されている21).
2)細胞膜ダイナミクス
Arf6は,神経突起伸長や細胞遊走に必須の細胞膜構造体であるラッフル膜の形成など,細胞膜ダイナミクスを伴うさまざまな細胞現象に関与していることが示されている22–24).これらの現象では,Arf6が制御するアクチン細胞骨格の再構成が重要な役割を果たす.アクチン細胞骨格の再構成において,Arf6-PIP5Kシグナル系は重要な役割を担っている.ラッフル膜の形成過程では,Arf6は細胞辺縁部でPIP5Kを活性化してPI(4,5)P2の産生を誘導する22).PI(4,5)P2は,さまざまなアクチン結合タンパク質と結合してそれらの活性を制御するため,Arf6-PIP5Kシグナル系の下流ではアクチン細胞骨格のダイナミックな変動が誘起されて,細胞辺縁部でのラッフル膜の形成が誘導される.また,低分子量Gタンパク質Rac1はアクチン細胞骨格の再構成を制御する重要な分子であるが,Arf6の下流ではRac1が機能し,細胞遊走を制御していることが示されている25, 26).しかし,我々はArf6の上流においてもRac1が機能している結果を得ており22),Arf6とRac1のシグナル伝達における位置関係は単純ではないと考えられる.
インベイドポディア(浸潤仮足)は,がん細胞の浸潤に必須の細胞膜構造体であるが,Arf6はGAPの一つであるAMAP1との結合を介してインベイドポディア形成を制御する9).AMAP1はアクチン細胞骨格結合タンパク質であるコータクチンや細胞接着関連分子パキシリンと結合して,これらのタンパク質を細胞膜へリクルートすることによりインベイドポディア形成を促進すると考えられる27).また,がん細胞が浸潤するためには,周囲の細胞外マトリックスの分解が必要であるが,Arf6は膜結合型マトリックスメタロプロテアーゼ1を含んだ微小胞の放出に関与していることが報告されている28).微小胞の放出とは,細胞膜が外側に隆起してくびり切れる現象であり,この現象においては,Arf6の下流分子としてPLDが機能していることが示されている.
上述したように,Arf6の細胞レベルでの生理機能は精力的に解析されてきており,多くの知見が蓄積しつつある.Arf6の制御する細胞内小胞輸送やアクチン細胞骨格の再構成は,細胞や組織の恒常性維持やさまざまな生体反応においてきわめて重要であるため,すべからくArf6は個体レベルにおいても重要な機能を担っていることが予想される.我々は,Arf6 KOマウス,および組織・細胞特異的にArf6遺伝子を欠失させたArf6コンディショナルノックアウト(cKO)マウスを作製し,個体レベルでのArf6の機能解析を進めている.以下に,我々の解析から得られた知見を含めて,個体レベルでのArf6の生理機能を概説する.
1)Arf6は肝臓の発生に必須である
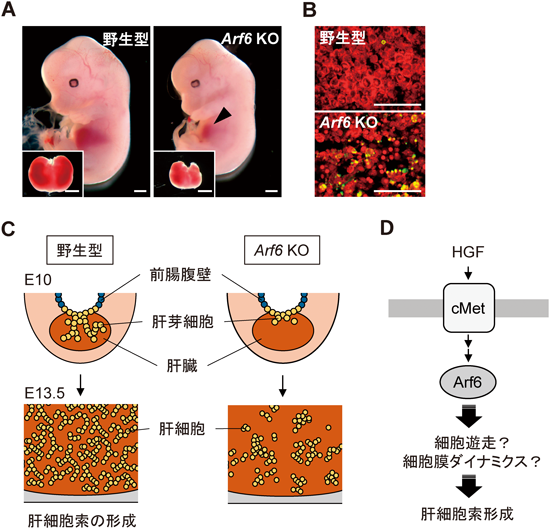
Arf6 KOマウスは,肝臓の発達不全を呈し,胎生中期から後期にかけて致死となる29).Arf6 KOマウスの肝臓は,野生型と比べてそのサイズが顕著に小さく(図2A),肝臓全体においてアポトーシスの亢進が確認される(図2B).細胞レベルでのArf6の機能が明らかになりつつある中で,このような知見は,Arf6 KOマウスを用いた解析でしか知りえないものであった.図2Cに示すように,肝臓は,胎生中期に肝芽細胞が前腸腹壁より周囲の間充織に浸潤し,そこで肝細胞に分化することで形成される.分化した肝細胞はその後,肝細胞索と呼ばれる網目状構造を形成し,肝臓の発達に寄与している.肝細胞索は,肝臓自体の構造を支持すると同時に,造血系細胞や間質系細胞との細胞間クロストークを介して,これらの細胞の生存に貢献している.マウス胎仔から摘出した肝臓の組織学的解析によると,Arf6 KOマウスでは胎生10日目における肝芽細胞の遊走が悪く,その後の肝臓における肝細胞索の形成が著しく低下していた(図2C).このような肝細胞の挙動は肝細胞増殖因子(hepatocyte growth factor:HGF)によって制御されている.in vitro三次元培養系において,初代肝細胞をHGFで刺激することによって肝細胞索様構造の形成が誘導できるが,Arf6 KO初代培養肝細胞においてはHGF刺激による肝細胞索様構造の形成が有意に低下していた.肝細胞索様構造の形成には,細胞遊走と細胞膜ダイナミクスが不可欠であり,おそらくArf6の欠失によってHGF依存的な細胞遊走や細胞膜ダイナミクスが障害されるために肝細胞索形成不全となり,その結果,肝臓でのアポトーシスの亢進,肝臓の発達不全につながっていることが想定される(図2D).
2)Arf6は中枢神経においてミエリン鞘形成を制御する
上述したように,Arf6 KOマウスは肝臓の発達異常により胎生中期から後期にかけて致死となる.このことは,in vivoにおけるArf6の重要性を如実に示す結果であったが,同時に個体におけるさらなるArf6の機能解析を困難にさせるものであった.そこで我々は,Cre-loxPシステムを利用したコンディショナルノックアウト法を用いて,特定の組織・細胞でArf6遺伝子を欠失させたArf6 cKOマウスを作製し,その解析を進めている.
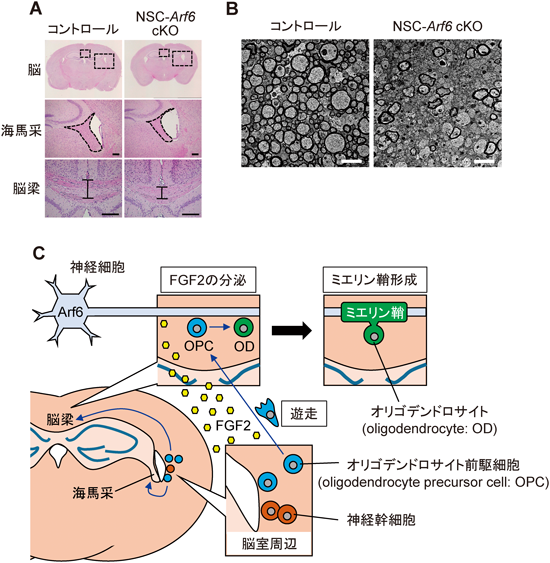
これまでの我々の解析および他グループの報告から,Arf6は脳に発現していることが知られていたため30, 31),我々は神経幹細胞においてCreを発現するNestin-Creマウスを用いて,脳特異的にArf6をノックアウトしたマウス(NSC-Arf6 cKOマウス)を作製してその解析を行った32).このマウスは一見して正常に発生・成長し,繁殖行動も正常であった.しかし興味深いことに,脳の組織学的解析から,このマウスの脳はコントロールマウスに比べてそのサイズがわずかに小さく,脳梁や海馬采の厚みが顕著に減少していた(図3A).脳梁および海馬采にはそれぞれ,左右の大脳半球間,海馬–乳頭体間をつなぐ神経線維が走行している.そのため,これらの領域の神経軸索は,オリゴデンドロサイトにより高度にミエリン化されており,上述の領域間で情報が速やかに伝達されるよう構成されている.そこで,電子顕微鏡解析によりこれらの領域の神経軸索ミエリン鞘の形成を観察したところ,NSC-Arf6 cKOマウスではミエリン化された軸索の割合が大幅に減少しており,それに伴い神経軸索の直径が有意に低下していることが明らかとなった(図3B).Arf6は神経軸索の伸長に関与することが報告されているが,少なくとも脳梁や海馬采においては神経軸索の密度はむしろ増加傾向を示していた.そのため,NSC-Arf6 cKOマウスに観察される脳梁や海馬采のサイズの減少は,神経軸索数の減少によるものではなく,ミエリン化の障害によるものであると結論づけられる.
脳梁や海馬采において,ミエリン鞘はオリゴデンドロサイトにより形成される.そこでこれらの領域におけるオリゴデンドロサイトの数を比較したところ,NSC-Arf6 cKOマウスでは,オリゴデンドロサイトの数が顕著に低下していた.図3Cに示すように,脳梁や海馬采に存在するオリゴデンドロサイトは,脳室周辺に存在する神経幹細胞に由来している.脳室周辺で神経幹細胞から分化したオリゴデンドロサイト前駆細胞は,脳梁や海馬采に遊走し,そこでオリゴデンドロサイトに終末分化した後に神経軸索をミエリン化する.また,このオリゴデンドロサイト前駆細胞の遊走は,神経細胞などからのガイダンス因子の放出によって制御されている33).そこで,神経細胞とオリゴデンドロサイトのどちらの細胞でArf6が機能しているのかを検討するために,Tau-Creを用いて神経細胞特異的にArf6をノックアウトしたマウス(N-Arf6 cKOマウス),および2′,3′-cyclic nucleotide 3″-phosphodiesterase(CNP)-Creを用いてオリゴデンドロサイト特異的にArf6をノックアウトしたマウス(O-Arf6 cKOマウス)を作製し,それぞれのマウスにおけるミエリン鞘の形成を検討した.興味深いことに,N-Arf6 cKOマウスでは脳梁,海馬采におけるミエリン鞘形成の障害が観察されたが,O-Arf6 cKOマウスではこの表現型は再現されなかった.すなわちこの結果は,神経細胞に発現するArf6がオリゴデンドロサイトのリクルートに関与するガイダンス因子の放出に寄与していることを示している.実際,N-Arf6 cKOマウスより摘出した海馬において,オリゴデンドロサイトのガイダンス因子である繊維芽細胞増殖因子2(fibroblast growth factor 2:FGF2)の分泌が有意に抑制されていた.以上のことから,神経細胞に発現するArf6は,FGF2の分泌を制御することによって脳梁および海馬采へのオリゴデンドロサイトのリクルートに関わっており,それゆえ,これらの領域における神経軸索のミエリン化に寄与していると考えられる(図3C).
3)Arf6は腫瘍血管新生を制御する
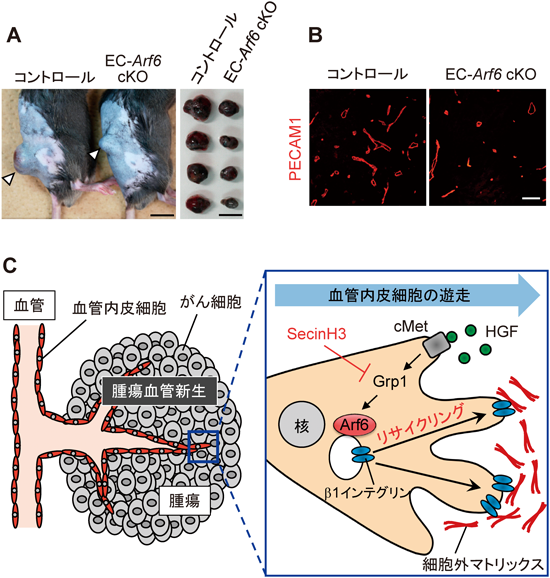
腫瘍組織を構成するがん細胞や,繊維芽細胞などのストローマ細胞は,血管内皮細胞増殖因子(vascular endothelial cell growth factor:VEGF)やFGF2, HGFなどのさまざまな血管誘導因子を分泌して周囲の既存血管を刺激し,腫瘍内に進入する腫瘍血管新生を誘導する34).腫瘍組織内に新生された血管は,腫瘍組織に酸素や栄養素を供給し,腫瘍組織の成長を助長する.1971年にJudah Folkmanにより「腫瘍血管新生の阻害による腫瘍への兵糧攻め」の概念が提唱されたことにより35),腫瘍血管新生の分子メカニズムの解明はがん研究領域において重要な研究対象となり,その試みが精力的に行われてきた.興味深いことに,血管内皮細胞特異的なArf6 cKOマウス(EC-Arf6 cKOマウス)の皮下にB16メラノーマ細胞あるいはLewis肺がん細胞を移植して腫瘍の増大を経時的に観察したところ,EC-Arf6 cKOマウスではコントロールマウスと比較して腫瘍の成長が抑制され(図4A),腫瘍血管新生の顕著な阻害が観察された36)(図4B).この結果は,血管内皮細胞に発現するArf6が腫瘍血管新生を制御する重要な因子であることを示している.我々はArf6 KO血管内皮細胞を用いて,in vitroにおけるチューブ形成実験や大動脈リングアッセイによりVEGFやFGF2, HGFなどの血管誘導因子で促進される血管新生を解析したところ,Arf6の欠損はHGF依存的な血管新生のみを顕著に抑制することが明らかとなった.
血管新生には血管内皮細胞の遊走が必要であり,血管内皮細胞が遊走する際には,細胞外マトリックスとの適切な接着が必要である37).また,Arf6はこれまでに,接着分子であるβ1インテグリンの細胞内小胞輸送に関与することが報告されている16).そこで,血管内皮細胞におけるβ1インテグリンの細胞膜へのリサイクリングを解析したところ,Arf6 KO血管内皮細胞ではHGF依存的なβ1インテグリンのリサイクリングがほぼ完全に抑制されており,その結果,HGFによって誘導される血管内皮細胞の接着,遊走が阻害されていた.VEGF刺激によってもβ1インテグリンはエンドソームから細胞膜へリサイクリングされるが,興味深いことに,Arf6 KO血管内皮細胞ではVEGF依存的なβ1インテグリンのリサイクリングは阻害されない.このことは,VEGFシグナルとHGFシグナルではβ1インテグリンのリサイクリングシステムが異なっていることを示唆しており,Arf6はHGFシグナルが誘導するリサイクリングシステムにおいて重要であると結論づけられる.
Arf6がβ1インテグリンのリサイクリングを駆動するためには,アゴニスト刺激に伴ってArf6が活性化されることが必要であり,実際,HGF刺激によって血管内皮細胞のArf6は顕著に活性化される.このことは,HGFシグナルの下流でArf6 GEFが機能していることを意味している.上述のように,現在Arf6のGEFとしてEFA6ファミリー,BRAGファミリー,cytohesinファミリーの三つのファミリーが同定されている.これらのうち,cytohesinファミリーに属するArf6 GEFは,合成化合物SecinH3によってそのGEF活性が阻害されることが報告されている38).そこで我々は,SecinH3の存在下でβ1インテグリンのリサイクリングに対するSecinH3の影響を検討したところ,HGF依存的なβ1インテグリンのリサイクリングはSecinH3の添加によって顕著に抑制された.また,shRNAによるcytohesin分子のノックダウン解析から,cytohesin3/Grp1がHGF依存的なβ1インテグリンのリサイクリングに重要であることが明らかとなった.さらに,SecinH3はin vitroにおけるHGF依存的な血管新生,および個体における腫瘍血管新生を有意に抑制し,さらには,in vivoでの腫瘍の成長を阻害する効果を有していることが確認された.すなわち,血管内皮細胞においてGrp1-Arf6シグナル伝達系はHGF依存的なβ1インテグリンのリサイクリングを制御する重要なシグナル伝達系として位置づけることができ,このシグナル伝達系を抑制することによって腫瘍血管新生を抑制することが可能である(図4C).
4)Arf6シグナル伝達系の阻害とインスリン抵抗性
我々のArf6 KOおよびcKOマウスを用いた解析の他にも,これまでにcytohesin阻害剤SecinH3を使用してArf6の個体レベルでの生理機能に関する報告がなされている38).興味深いことに,SecinH3を投与したマウスはインスリンに対する抵抗性が亢進することが示されている.インスリンは肝臓でのグリコーゲン合成を促すが,SecinH3を投与したマウスの肝臓ではグリコーゲンの貯蔵レベルがコントロールマウスと比較して有意に減少する.また,マウスへのインスリン投与時には,肝臓において糖新生関連遺伝子の発現抑制と解糖関連遺伝子の発現上昇が観察されるが,これらの遺伝子群の発現変動はSecinH3の投与によって抑制される.これらのことから,SecinH3は肝臓におけるインスリンシグナル伝達系に対して抑制的に働き,インスリン抵抗性を誘導すると考えられる.実際,ヒト肝細胞株HepG2細胞をSecinH3で処理した際には,インスリン刺激依存的なAktの活性化やその下流で起こる遺伝子発現の変動が抑制される.SecinH3の薬理標的であるcytohesinファミリーには,cytohesin1~4の四つのアイソフォームが存在するが,このうちcytohesin2/ARNOとcytohesin3/Grp1の両方がインスリンシグナルにおいて重要であることが示されている.さらに,ARNOおよびGrp1は,Arf1とArf6の両方に対してGEF活性を有しているが,Arf1をノックダウンしたHepG2細胞ではインスリンシグナルは正常に機能するものの,Arf6をノックダウンした際には,インスリン刺激依存的な遺伝子発現の変動が抑制される.すなわち,インスリン受容体の下流ではcytohesin-Arf6シグナル伝達系が重要な役割を果たしていると考えられる.このシグナル伝達経路において,Arf6がどのような分子機構によってインスリンシグナルを制御しているのかは不明な点が多い.しかし,ARNO, Grp1, Arf6はともにインスリン刺激依存的にインスリン受容体と複合体を形成することが示されており38),インスリンシグナル伝達経路のきわめて初期の段階を制御しているようである.insulin receptor substrate (IRS)は,活性化したインスリン受容体と結合し,phosphatidylinositol 3-kinaseにシグナルを伝達するインスリン受容体の重要なアダプタータンパク質である.興味深いことに,SecinH3は,IRSとインスリン受容体との結合を阻害することが示されている38).すなわち,cytohesin-Arf6シグナル伝達系はインスリン受容体と結合することにより,IRSとインスリン受容体との結合を補助することによって,インスリンシグナルを正に制御していると考えられる.しかし受容体直下で活性化されたArf6が,どのような分子機構でIRSとインスリン受容体との結合を誘導するのかは不明であり,その詳細の解明は今後の解析に期待される.
Arf6 KOおよびArf6 cKOマウスの作出によって,個体レベルでのArf6の生理機能に関する知見が蓄積しつつある.しかし,その解析は始まったばかりであり,今後は個体レベルでさまざまな組織,細胞におけるArf6の生理機能が明らかにされていくことが期待される.Arf6はマウス個体においてユビキタスに発現する生命分子であり,その機能の多様性はまだまだ広がりを予想させる.我々はin situハイブリダイゼーション法を用いてマウス個体におけるArf6の発現解析を行い,皮膚や胃,肺,小腸,腎臓などのさまざまな組織の上皮細胞にArf6 mRNAが高発現していることを示している30).これらの組織におけるArf6の機能解析は今後の課題として残されている.
本稿で述べたように,Arf6は腫瘍血管新生において重要な役割を果たして,腫瘍形成を制御することが明らかとなり,Arf6シグナル伝達系を標的とした抗がん剤の開発も期待できよう.現在,腫瘍血管新生を阻害する薬剤としてスニチニブやベバシズマブなどのVEGFシグナルを標的とした血管新生阻害剤が開発され,がん患者の治療に応用されてきている.しかし,これらの腫瘍血管新生阻害剤は患者の寿命を数か月延長する効果をもたらすにすぎず,腫瘍血管新生阻害剤の新たな開発,あるいはその改良は現在も大きな課題として残っている39).近年,腫瘍血管新生阻害剤を用いた治療の危険性として,腫瘍血管新生を阻害することでがん細胞の転移能が亢進されることが示されている40, 41).これは腫瘍血管新生の阻害に伴ってがん細胞の低酸素シグナルが活性化されるためと考えられている.このような知見から,腫瘍血管新生阻害剤を治療に用いる場合は,がん細胞の転移能も平行して抑制する必要があることを意味している.これまでに,Arf6はがん細胞のインベイドポディア形成や,E-カドヘリンのエンドサイトーシス,微小胞放出等に関与し,がん細胞の転移・浸潤を正に制御していることが報告されている13, 27, 28).これらの報告と,上述した血管内皮細胞におけるArf6の機能を考え合わせると,Arf6の阻害薬を開発すれば,腫瘍血管新生を阻害すると同時にがん細胞の転移・浸潤を抑制できるはずである.
今後Arf6の個体レベルでの機能解析が進み,各種疾患との関係性がよりいっそう明確になっていくと予想している.各種疾患におけるArf6の分子機能や,GEF, GAP,エフェクター分子などのArf6シグナル伝達系の全体像が明らかになり,Arf6シグナル伝達系の創薬標的としての可能性が大きく広がることを期待したい.
引用文献References
1) Tsuchiya, M., Price, S.R., Tsai, S.C., Moss, J., & Vaughan, M. (1991) J. Biol. Chem., 266, 2772–2777.
2) D’Souza-Schorey, C. & Chavrier, P. (2006) Nat. Rev. Mol. Cell Biol., 7, 347–358.
3) Hongu, T. & Kanaho, Y. (2014) Adv. Biol. Regul., 54, 59–66.
4) Randazzo, P.A., Inoue, H., & Bharti, S. (2007) Biol. Cell, 99, 583–600.
5) Tanabe, K., Torii, T., Natsume, W., Braesch-Andersen, S., Watanabe, T., & Satake, M. (2005) Mol. Biol. Cell, 16, 1617–1628.
6) Natsume, W., Tanabe, K., Kon, S., Yoshida, N., Watanabe, T., Torii, T., & Satake, M. (2006) Mol. Biol. Cell, 17, 2592–2603.
7) Perez-Mansilla, B., Ha, V.L., Justin, N., Wilkins, A.J., Carpenter, C.L., & Thomas, G.M. (2006) Biochim. Biophys. Acta, 1761, 1429–1442.
8) Li, J., Ballif, B.A., Powelka, A.M., Dai, J., Gygi, S.P., & Hsu, V.W. (2005) Dev. Cell, 9, 663–673.
9) Onodera, Y., Hashimoto, S., Hashimoto, A., Morishige, M., Mazaki, Y., Yamada, A., Ogawa, E., Adachi, M., Sakurai, T., Manabe, T., Wada, H., Matsuura, N., & Sabe, H. (2005) EMBO J., 24, 963–973.
10) Inoue, H. & Randazzo, P.A. (2007) Traffic, 8, 1465–1475.
11) Claing, A., Chen, W., Miller, W.E., Vitale, N., Moss, J., Premont, R.T., & Lefkowitz, R.J. (2001) J. Biol. Chem., 276, 42509–42513.
12) D’Souza-Schorey, C., Li, G., Colombo, M.I., & Stahl, P.D. (1995) Science, 267, 1175–1178.
13) Palacios, F., Schweitzer, J.K., Boshans, R.L., & D’Souza-Schorey, C. (2002) Nat. Cell Biol., 4, 929–936.
14) Wenk, M.R. & De Camilli, P. (2004) Proc. Natl. Acad. Sci. USA, 101, 8262–8269.
15) Poupart, M.E., Fessart, D., Cotton, M., Laporte, S.A., & Claing, A. (2007) Cell. Signal., 19, 2370–2378.
16) Powelka, A.M., Sun, J., Li, J., Gao, M., Shaw, L.M., Sonnenberg, A., & Hsu, V.W. (2004) Traffic, 5, 20–36.
17) Li, J., Peters, P.J., Bai, M., Dai, J., Bos, E., Kirchhausen, T., Kandror, K.V., & Hsu, V.W. (2007) J. Cell Biol., 178, 453–464.
18) Zimmermann, P., Zhang, Z., Degeest, G., Mortier, E., Leenaerts, I., Coomans, C., Schulz, J., N’Kuli, F., Courtoy, P.J., & David, G. (2005) Dev. Cell, 9, 377–388.
19) Prigent, M., Dubois, T., Raposo, G., Derrien, V., Tenza, D., Rosse, C., Camonis, J., & Chavrier, P. (2003) J. Cell Biol., 163, 1111–1121.
20) Niedergang, F., Colucci-Guyon, E., Dubois, T., Raposo, G., & Chavrier, P. (2003) J. Cell Biol., 161, 1143–1150.
21) Grant, B.D. & Donaldson, J.G. (2009) Nat. Rev. Mol. Cell Biol., 10, 597–608.
22) Honda, A., Nogami, M., Yokozeki, T., Yamazaki, M., Nakamura, H., Watanabe, H., Kawamoto, K., Nakayama, K., Morris, A.J., Frohman, M.A., & Kanaho, Y. (1999) Cell, 99, 521–532.
23) Albertinazzi, C., Za, L., Paris, S., & de Curtis, I. (2003) Mol. Biol. Cell, 14, 1295–1307.
24) Santy, L.C. & Casanova, J.E. (2001) J. Cell Biol., 154, 599–610.
25) Hu, B., Shi, B., Jarzynka, M.J., Yiin, J.J., D’Souza-Schorey, C., & Cheng, S.Y. (2009) Cancer Res., 69, 794–801.
26) Santy, L.C., Ravichandran, K.S., & Casanova, J.E. (2005) Curr. Biol., 15, 1749–1754.
27) Sabe, H., Hashimoto, S., Morishige, M., Ogawa, E., Hashimoto, A., Nam, J.M., Miura, K., Yano, H., & Onodera, Y. (2009) Traffic, 10, 982–993.
28) Muralidharan-Chari, V., Clancy, J., Plou, C., Romao, M., Chavrier, P., Raposo, G., & D’Souza-Schorey, C. (2009) Curr. Biol., 19, 1875–1885.
29) Suzuki, T., Kanai, Y., Hara, T., Sasaki, J., Sasaki, T., Kohara, M., Maehama, T., Taya, C., Shitara, H., Yonekawa, H., Frohman, M.A., Yokozeki, T., & Kanaho, Y. (2006) Mol. Cell. Biol., 26, 6149–6156.
30) Akiyama, M., Zhou, M., Sugimoto, R., Hongu, T., Furuya, M., Funakoshi, Y., Kato, M., Hasegawa, H., & Kanaho, Y. (2010) Dev. Dyn., 239, 3416–3435.
31) Matsuya, S., Sakagami, H., Tohgo, A., Owada, Y., Shin, H.W., Takeshima, H., Nakayama, K., Kokubun, S., & Kondo, H. (2005) J. Neurochem., 93, 674–685.
32) Akiyama, M., Hasegawa, H., Hongu, T., Frohman, M.A., Harada, A., Sakagami, H., & Kanaho, Y. (2014) Nat. Commun., 5, 4744.
33) de Castro, F. & Bribian, A. (2005) Brain Res. Rev., 49, 227–241.
34) Montag, M., Dyckhoff, G., Lohr, J., Helmke, B.M., Herrmann, E., Plinkert, P.K., & Herold-Mende, C. (2009) Cancer Sci., 100, 1210–1218.
35) Folkman, J. (1971) N. Engl. J. Med., 285, 1182–1186.
36) Hongu, T., Funakoshi, Y., Fukuhara, S., Suzuki, T., Sakimoto, S., Takakura, N., Ema, M., Takahashi, S., Itoh, S., Kato, M., Hasegawa, H., Mochizuki, N., & Kanaho, Y. (2015) Nat. Commun., 6, 7925.
37) Silva, R., D’Amico, G., Hodivala-Dilke, K.M., & Reynolds, L.E. (2008) Arterioscler. Thromb. Vasc. Biol., 28, 1703–1713.
38) Hafner, M., Schmitz, A., Grune, I., Srivatsan, S.G., Paul, B., Kolanus, W., Quast, T., Kremmer, E., Bauer, I., & Famulok, M. (2006) Nature, 444, 941–944.
39) Mitchell, D.C. & Bryan, B.A. (2010) J. Cell. Biochem., 111, 543–553.
40) Ebos, J.M., Lee, C.R., Cruz-Munoz, W., Bjarnason, G.A., Christensen, J.G., & Kerbel, R.S. (2009) Cancer Cell, 15, 232–239.
41) Paez-Ribes, M., Allen, E., Hudock, J., Takeda, T., Okuyama, H., Vinals, F., Inoue, M., Bergers, G., Hanahan, D., & Casanovas, O. (2009) Cancer Cell, 15, 220–231.
著者紹介Author Profile
 本宮 綱記(ほんぐう つなき)
本宮 綱記(ほんぐう つなき)筑波大学国際テニュアトラックプログラム(医学医療系)助教.博士(医学).
略歴1981年愛媛県に生る.2004年神奈川大学理学部卒業.06年東京都立大学大学院理学研究科修士課程修了.11年筑波大学大学院人間総合科学研究科博士課程修了.12年同大学グローバル教育院ヒューマンバイオロジー学位プログラム(医学医療系)助教.14年より現職.
研究テーマと抱負低分子量G蛋白質Arf6の活性調節機構および個体レベルでの生理機能解析.複雑なArf6シグナル伝達系の全貌を,分子,個体の両面から理解していきたいと考えています.
趣味映画鑑賞,子供と遊ぶこと.
 金保 安則(かなほ やすのり)
金保 安則(かなほ やすのり)筑波大学医学医療系教授.薬学博士.
略歴1953年京都府に生る.76年京都薬科大学薬学部卒業.83~89年アメリカ留学(NIH他)後,岐阜大学医学部,東京工業大学生命理工学部,東京都臨床医学総合研究所を経て,2005年より現職.
研究テーマと抱負個体レベルでの低分子量G蛋白質Arf6の生理機能の全貌を解明し,Arf6の病態における役割を明らかにすることにより,創薬研究開発の基盤を構築したい.
ウェブサイトhttp://www.md.tsukuba.ac.jp/basic-med/biochem/kanaholab/index.html
趣味スキー,筋トレ.