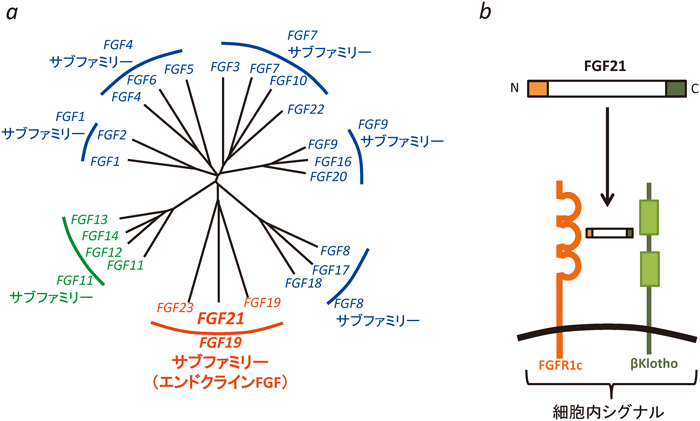
線維芽細胞増殖因子(fibroblast growth factor:FGF)は,線維芽細胞をはじめとするさまざまな細胞に対して増殖活性や分化誘導など多彩な作用を示す多機能性細胞間シグナル因子である1).FGFは,ヒトやマウスにおいて22種類存在する.この22種類のFGFは,アミノ酸配列の相同性から,さらに七つのサブファミリーに分類される(図1a)1).そのうち五つのサブファミリーに属する15種類のFGFに関しては,いわゆる傍分泌因子として機能し(パラクラインFGF),4種類のFGF(FGF11~14)は,そのN末端に分泌性シグナル配列を持たず,細胞内で作用することが明らかにされている(イントラクラインFGF).そして,残りの3種類のFGF(FGF19, FGF21, FGF23)は,内分泌因子としても作用しうることから,エンドクラインFGFと呼ばれている.3種類のエンドクラインFGFは,いずれも個体の代謝調節に重要な役割を果たすが,その中でも,FGF21は,肥満症や肥満症を発症基盤とする代謝異常に対する治療薬の候補因子として注目されている.本稿では,このFGF21の構造,薬理作用,生理的意義などについて紹介したい.
FGF21は,FGF19の配列との相同性をもとに,マウス胎仔cDNAより単離された2).ヒトの染色体では19q13.33に位置しており,四つのエクソンから構成されている.その中で,コーディング領域は第2エクソンから始まり,第4エクソン途中で終了する.ヒトFGF21は209アミノ酸残基を,マウスFGF21は210アミノ酸をコードしており,いずれもそのN末端には28アミノ酸残基からなる典型的なシグナル配列が存在する.FGF21を除くほとんどのFGFでは,そのC末端側にヘパリンやヘパラン硫酸に結合する部位が存在する.一方,FGF21を含むエンドクラインFGFではその部分の構造が異なり,細胞周囲に発現するヘパラン硫酸との親和性が低い.この親和性の低さが,エンドクラインFGFが産生組織にとどまらず遠方へ拡散する一つの理由であると考えられている3).
FGF21は,他の多くのFGFと同様に,細胞表面に存在するチロシンキナーゼ型受容体のFGF受容体(FGFR)に結合することにより活性を示す1).FGFR遺伝子は,ゲノム上に1~4の4種類存在しており,さらに,FGFR1~3については,bタイプとcタイプの2種類のスプライシングバリアントが存在することが知られている.in vitroの結合実験や,脂肪組織においてFGFR1を欠損するマウスにおいてFGF21の多くの薬理作用が消失あるいは減弱することなどから,生体におけるFGF21の主要な受容体はFGFR1cであると考えられている4).また,FGF21においては,膜貫通型の糖タンパク質であるKlothoファミリーが共受容体として機能することが知られている1).Klothoファミリーが共受容体となるのは,三つのエンドクラインFGFに共通しており,FGF19やFGF21はβKlotho分子を,FGF23はαKlotho分子を共受容体とする.実際に,FGF21の作用におけるβKlotho分子の必要性は,in vitroの解析だけではなく,全身および脂肪組織特異的なβKlothoノックアウトマウスを用いても明らかにされている5).FGF21と受容体,共受容体の複合体形成に関しては,FGF21のN末端部分がFGFR1cと,C末端部分がβKlothoと結合することが明らかになっている(図1b)6).
3. FGF21の有する薬理作用とその臨床応用に関して
FGF21が発見された当時,FGFは基本的に産生細胞の近傍で作用すると考えられていた.したがって,肝臓に発現するFGF21の生理的意義について,我々は肝臓での役割を中心に検討していたが,ほとんど成果は得られていなかった.その一方で,2005年,FGF21に関する非常に重要な知見が報告された7).Kharitonenkovらは,培養ヒト白色脂肪細胞において糖取り込みを促進する因子を探索し,FGF21がその活性を有することを見いだしたのである.さらに,過剰発現マウスの解析や,肥満動物への投与実験などを行い,FGF21にインスリン感受性の改善や,血中トリアシルグリセロール(トリグリセリド)濃度の低下,体重増加の抑制などの作用があることを報告した.その後,FGF21の投与により,全身レベルでの脂質利用能の亢進,エネルギー消費の亢進,肝臓におけるトリグリセリド蓄積の抑制や脂肪酸酸化などが起こることや,糖尿病を発症したサルに対する投与で,糖代謝などが改善することなど,FGF21の持つ薬理作用が次々と報告された8, 9).
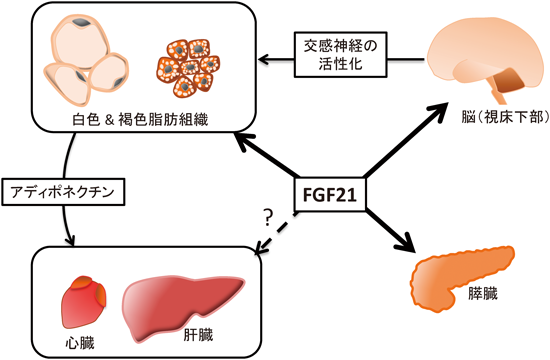
これまでの肥満症,糖代謝異常や脂質代謝異常に対する薬品では,それぞれの症状に個別に対処することはできるものの,すべての症状に対し効果を示すものは少ない.それに対し,FGF21は,それ一つでさまざまな代謝異常を改善しうることが大きな特徴である.さらに,同じエンドクラインFGFの一つFGF19は,肝細胞増殖,がん化を起こすのに対し,FGF21は明確な細胞増殖活性を持たず,個体への投与実験においてもがんの発生は確認されていない7).加えて,FGF21は,過剰投与による低血糖が起こらない9).これらの特徴を持つFGF21は,肥満や肥満症由来の疾患治療薬の候補因子として期待されている.では,FGF21の代謝改善作用は,どのようなメカニズムで起こるものであろうか.以下では,FGF21の薬理作用の標的となる臓器について,簡単にまとめてみた(図2参照).
1)FGF21の脂肪組織に対する作用
代謝調節臓器の一つである脂肪組織は,薬理作用が発見された当時からFGF21の作用臓器として考えられていた.この脂肪組織であるが,生体には主に余剰エネルギーの保管庫である白色脂肪組織と余剰エネルギーを熱として放散する褐色脂肪組織が存在する.白色脂肪組織の構成細胞である白色脂肪細胞では,余剰エネルギーを中性脂肪の形で細胞内に蓄積し,また必要に応じて脂肪酸とグリセロールに脂肪分解して,血中へ放出する.一方,褐色脂肪組織における褐色脂肪細胞では,脂肪酸のβ酸化により得られるはずのエネルギーを,ミトコンドリアに存在する脱共役タンパク質1(UCP1)を用いて,熱として放散してしまう.先に述べたように,FGF21は白色脂肪細胞において糖取り込みを促進したことから,直接の標的細胞として白色脂肪細胞が予想された7).実際,脂肪細胞特異的なFGFR1ノックアウトマウスにFGF21を投与したところ,FGF21の有する糖代謝,エネルギー消費促進作用のほとんどが消失し,また,βKlothoを脂肪細胞特異的にノックアウトしたマウスにFGF21を投与したところ,糖代謝改善作用が消失した4, 5).以上より,FGF21による代謝調節には,脂肪細胞に対する直接的な作用が重要であることが示された.
一方,脂肪組織以外の臓器に対しては,FGF21が脂肪組織を介して間接的に作用する可能性が考えられる.我々は,この間接的な作用に脂肪細胞由来の内分泌因子であるアディポネクチンが重要であることを明らかにした10).アディポネクチンは肝臓や筋肉などに直接作用して,インスリン感受性の低下,脂質代謝異常や脂肪肝などを改善する.FGF21の投与により,脂肪細胞より分泌されるアディポネクチン量が増加した.また,アディポネクチンノックアウトマウスにFGF21を投与すると,FGF21の持つ糖代謝改善作用,脂質代謝改善作用,エネルギー消費増加などの作用が減弱した.これらの結果は,FGF21の薬理作用において,脂肪細胞由来のアディポネクチンが他臓器への間接的な作用に重要であることを示している.また,最近,心筋梗塞モデルマウスに対するFGF21の投与により,梗塞部位の減少が起こることが報告され,これも脂肪組織から産生されるアディポネクチンを介した間接的な作用であることが明らかにされた11).
一方,二つの脂肪組織の機能は,交感神経刺激により調節されている.交感神経末端から放出されるノルアドレナリンにより,褐色脂肪組織の脂肪分解や脂肪酸酸化,熱産生は促進し,白色脂肪細胞では脂肪分解が誘導される.また,交感神経刺激,あるいは寒冷曝露により,白色脂肪組織内にUCP1陽性の褐色脂肪細胞様の細胞(ベージュ細胞)が生じ,個体のエネルギー消費に寄与する.2012年,FGF21をマウスに投与することにより,褐色脂肪組織における脂肪酸酸化関連因子の発現やUCP-1などの発現が上昇し,また鼠蹊部白色脂肪組織においてベージュ細胞が出現することが報告された12).また,同報告では,初代培養細胞を用いて,この作用がFGF21の直接的な作用であることも示されている.したがって,FGF21は,褐色脂肪組織の活性化やベージュ細胞の誘導を介して,エネルギー消費を促し,体重を減少させる可能性が示唆される.
2)FGF21の膵臓,肝臓に対する作用
脂肪組織特異的FGFR1ノックアウトマウスにおいても,FGF21の投与直後に膵臓で転写因子Egr-1が誘導されたため,FGF21は膵臓にも直接作用する可能性がある4).実際,糖尿病モデルラットあるいは糖尿病モデルマウスの膵臓ランゲルハンス島にFGF21を添加すると,インスリン分泌の促進や含有するインスリン量が増加する13).また株化β細胞などを用いた検討では,FGF21は,糖脂肪毒性やサイトカインにより起こるアポトーシスを抑制する.これらの培養実験の結果に一致して,糖尿病モデルマウスにFGF21を持続投与することにより,ランゲルハンス島の数やランゲルハンス島におけるβ細胞の数が有意に増加することが報告されている.
では,肝臓への直接的な作用はどうだろうか.肥満モデルマウスへの投与において,FGF21は肝臓のトリグリセリドの蓄積を抑制し,脂肪酸酸化を誘導する.しかし,培養肝細胞への添加実験では,FGF21が肝細胞に直接作用する証拠はほとんど得られていない.また,FGFR1が生体の肝細胞にほとんど発現していないことも知られている14).したがって,FGF21が肝臓に直接作用する可能性は低いものと思われる.一方,ヒト肝がん由来細胞株のHepG2において,FGF21は脂肪酸合成やトリグリセリドの合成に関わる転写因子や,動脈硬化に関係するアポリポプロテイン(a)の発現を抑制することが報告されている15, 16).しかし,HepG2はFGFR1を高発現しており,FGFR1をほとんど発現していない生体の肝臓を正しく反映していない可能性に注意すべきである16).肝細胞に対する直接の作用の解析には,肝細胞特異的なβKlothoあるいはFGFR1ノックアウトマウスに対するFGF21の投与実験が重要になるが,これまで両マウスに関する報告はない.
3)FGF21の中枢神経系に対する作用
中枢神経系にFGF21の発現は認められないが,マウスにおいて末梢に投与されたFGF21が,一部中枢神経系に移行することが報告された17).さらに,中枢神経系にβKlothoが発現することも合わせると,皮下などに投与されたFGF21が,直接中枢神経系に作用している可能性が考えられる.この中枢神経系への直接作用を明らかにするために,神経系特異的なβKlothoノックアウトマウスの解析が行われた4, 18).その結果,FGF21の投与直後に起こる糖代謝の改善には関与しないものの,FGF21は視床下部への直接作用を介して交感神経系を活性化し,その結果,白色脂肪組織の褐色化,あるいは褐色脂肪組織の活性化,エネルギー消費の亢進などが起こることが明らかにされた.Dourisらも,FGF21によるエネルギー消費の亢進において,中枢神経系への作用に続く交感神経系の活性化が重要であることを指摘している19).またOwenらは,FGF21には,中枢への直接作用を介した交感神経系の活性化と,脂肪組織への直接的な作用である糖取り込みとβ酸化などの亢進の二つの薬理作用が存在し,その二つが組み合わさることで,効率よくエネルギー消費が促進されるのではないかと述べている18).
4)その他
最近では,炎症性の疾患に対するFGF21の投与効果に関する報告がいくつかみられる.腎臓の炎症がFGF21を投与することで改善すること,あるいは病原体感染によって起こる全身性炎症疾患である敗血症に関して,FGF21の投与により,敗血症モデルマウスの生存率が回復することが報告された20, 21).また,FGF21は,カタラーゼやスーパーオキシドジスムターゼの活性の上昇を通じて酸化ストレスを軽減し,また炎症反応を抑制することにより,関節リウマチモデルマウスの症状を改善することが報告された22).さらに,肥大心に対する薬理作用として,FGF21が直接心筋細胞における炎症や活性酸素の産生に対し抑制的に働くことが示唆されている23).炎症や活性酸素などは,代謝性疾患をはじめとするさまざまな疾患の発症基盤となるため,今後,FGF21の薬理作用のメカニズムを考える上で,重要なポイントになるかもしれない.
5)FGF21シグナリングの医療への応用
以上のように,魅力的な薬理作用を有するFGF21であるが,薬として利用する際には改善されるべき問題も存在する.まず問題となるのは,その半減期の短さである.投与する動物種や投与方法にもよるが,体内におけるFGF21の半減期は0.5時間から5時間程度しかない.また,nativeなFGF21は熱に対する安定性が低く,溶液中で凝集する傾向がある.そこで,FGF21にさまざまな修飾を加えることにより,それらの問題を解決する試みが行われている.たとえば,ポリエチレングリコール(PEG)による修飾があげられる.一般的に,PEG修飾された化合物は,腎臓における排泄が抑制され,免疫原性も低下する.131番目のアルギニン残基を標的にPEGを付加したPEG修飾FGF21については半減期が飛躍的に改善し,肥満糖尿病モデルマウスに対し1週間に2度投与するだけで,糖代謝や脂質代謝を改善した24).その他,ヒトFGF21のN末端側にIgGのFc領域を融合させることにより,半減期を延長する試みも行われている25).この試みでは,Fc領域を融合させるだけではなく,FGF21の分解を防ぐために171番目のプロリンをグリシンに,さらに高濃度で起こる凝集を防ぐために98番目のロイシンをアルギニンに置換した改変体Fc-FGF21(RG)を作成した.このFc-FGF21(RG)は,半減期がマウスで11時間,サルで30時間にまで延長しており,肥満症モデルマウスでは,5日間に1回の頻度で皮下投与することにより,血糖の低下や体重増加抑制作用が維持された.
以上のようなFGF21の改変体の中で,LY2405319はヒトを対象とした投与成績も報告されている.LY2405319は,ヒトFGF21をベースにして,N末端に起こる分解を防ぐためにN末端の四つのアミノ酸を除去し,また不均一性の原因となるO型糖鎖の付加を防ぐために167番目のセリン残基をアラニンに変更している26).また,熱安定性の向上と凝集の防止を目的に,118番目のロイシン残基と134番目のアラニン残基を二つともシステインに変更し,新規なジスルフィド結合を導入した.このLY2405319は,肥満モデル動物に対して一度皮下投与することにより,糖代謝改善作用や体重減少が14日程度持続した.また,肥満症患者や糖尿病患者に対して実際に投与したところ,1日1回最大20 mgの皮下投与を28日間続けると,LDL,総コレステロール,血中トリグリセリドが低下し,HDLの上昇が認められた27).さらに,有意ではないものの体重の減少も認められた.一方,空腹時血糖や血中インスリン濃度は低下したもののわずかであり,有意な効果は認められなかった.
ここまでに説明したもの以外にも,さまざまな分子が創出され,その有用性が報告されている28).たとえば,従来のFGF(FGF2など)のヘパリン/ヘパラン硫酸結合領域を除去し,そこにβKlothoとの結合が期待されるFGF21のC末端領域を融合する試みなどが行われている.また,アゴニスト活性を示す抗FGFR1c抗体を探索し,FGF21の代わりに使用する試みなどもある.しかし,いずれも動物実験レベルでの検討であり,今後はヒトへの投与を視野に入れていく必要があると思われる.
これまではFGF21の薬理作用を中心にまとめてきた.一方で,生体内で産生されるFGF21には生理的な意義も存在すると思われる.そこで,FGF21の生理的意義に関して,明らかにされた知見を以下にまとめる.
1)FGF21の発現および血中濃度の変化
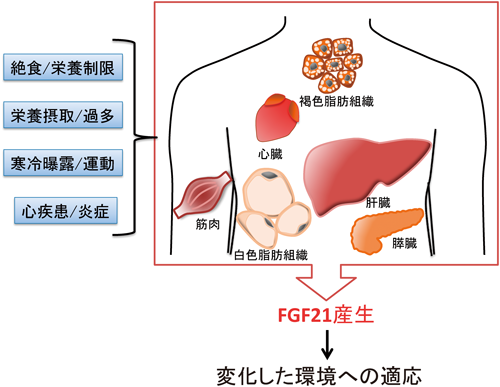
内分泌因子の生理的意義を考える上で,臓器における産生量や,血中量の変化は手がかりとなる.しかし,FGF21の発見からしばらくは,通常時の肝臓と胸腺以外の発現に関する情報はほとんどなかった2).しかし,2007年に,絶食や長期の飢餓状態を模倣するといわれる高脂肪低炭水化物食(ケトン食)飼育により,FGF21が肝臓で著しく発現誘導され,血中濃度が著しく上昇することが報告された29, 30).この報告以降,FGF21の発現や血中濃度が上昇する条件,あるいは脂肪組織や筋肉など肝臓以外の産生臓器に関する知見が次々と明らかにされていった(図3).以下,このFGF21の発現上昇,血中濃度増加に関わる刺激,あるいは外的要因をまとめる.
先に述べたように,肝臓におけるFGF21は,絶食やケトン食飼育時のような糖質の供給が著しく制限される条件で発現が誘導される.このFGF21の発現誘導には,peroxisome proliferator-activated receptor α (PPARα),cAMP-responsive element binding protein H(CREBH)といった転写因子や,細胞内の栄養センサーとして機能するNAD依存性脱アセチル化酵素Sirtuin1(Sirt1)などが関わる29, 31, 32).また,タンパク質やアミノ酸が不足した条件下においてもFGF21の発現が誘導される.たとえば,食餌中のタンパク質含有量を制限することにより,肝臓におけるFGF21の発現が誘導される33).その他,ロイシンを含まない餌でマウスを飼育すると,マウスの体重や脂肪量の増加が抑制されることが知られているが,このような栄養条件において,FGF21は,activating transcription factor 4(ATF4)依存性に,肝臓で発現が上昇する34).マウスの非アルコール性脂肪性肝炎(NASH)モデルを作製する際に利用されるメチオニン/コリン制限食でも,肝臓におけるFGF21の発現が誘導されることが報告されている35).
栄養素の摂取を制限した場合において発現が誘導される一方,逆に栄養素の摂取によってもFGF21の産生が増加することも知られている.たとえば,高脂肪高炭水化物食により肥満症を発症したマウスでは,肝臓や白色脂肪組織においてFGF21の発現が上昇し,FGF21の血中量が増加することが知られている.FGF21の薬理作用を考慮すると,肥満症発症時にFGF21の血中濃度が増加していることは矛盾を感じるが,この説明として,肥満症発症時にFGF21抵抗性が生じることが示唆されている36).また,最近では糖質であるフルクトースの摂取2時間後に,ヒト循環血中のFGF21濃度が急速に上昇することが報告された37).なお,同じ単糖のグルコースもcarbohydrate response element binding protein(ChREBP)を介してFGF21の産生を誘導するが,フルクトースの投与と比べ,グルコースの投与では緩やかにFGF21濃度が上昇した.また,我々も,ある種の食用油を経口投与することにより,FGF21の肝臓における発現,血中濃度が上昇することを見いだしている(未公開データ).現在,この食用油によるFGF21の誘導に関して,メカニズムや薬理的な意義に関して検討中である.
また,栄養摂取の変化以外にも内因性のFGF21の発現が上昇する条件がいくつか報告されている.たとえば,寒冷曝露刺激では,褐色脂肪組織におけるFGF21の産生が誘導される.寒冷曝露刺激時には交感神経系が活性化するが,実際,ノルアドレナリンやβ3アゴニストの投与によっても,p38α-ATF2経路によって褐色脂肪細胞におけるFGF21の産生は上昇した38).また,運動によっても,肝臓におけるFGF21の産生や血中濃度は上昇する39).実験的な敗血症モデルや膵炎モデルにおいても,それぞれ白色脂肪組織と筋肉,膵臓における発現が上昇することも報告されている21, 40).なお,敗血症モデルマウスでは,肝臓におけるFGF21の産生が減少するにも関わらず,血中FGF21濃度が上昇するため,筋肉や白色脂肪組織で産生されたFGF21が血中へ放出されている可能性が示唆されている.さらに,最近,病態心とFGF21との関わりについても明らかになりつつあるが,心肥大誘導時に,Sirt1, PPARαの経路により心筋細胞においてFGF21の発現が上昇することや,マウスを用いた心筋梗塞モデルにおいて心臓における発現が上昇することも報告されている41).
2)ノックアウトマウスを用いたFGF21の生理的意義の解析
FGF21の生理的意義を明らかにするためには,FGF21ノックアウトマウスの表現型が重要である.FGF21ノックアウトマウスは,筆者らを含め複数の機関において作製されたが,通常食飼育下では大きな表現型の変化を示さない42–44).Dutchakらは,FGF21ノックアウトマウスの白色脂肪細胞において,分化のマスターレギュレーターである転写因子PPARγの活性が抑制され,それに伴って脂肪含量や白色脂肪細胞のサイズが減少することを報告している45).しかし,他系統のFGF21ノックアウトマウスでは,どちらかというと体重や脂肪組織重量は,むしろ増加傾向である.
このような通常時の表現型解析に加えて,現在,FGF21の産生が上昇する条件下におけるFGF21ノックアウトマウスの表現型の解析が盛んに行われている.たとえば,絶食やケトン食負荷のような糖質の供給が著しく抑制される条件では,白色脂肪組織における脂肪分解により産生された脂肪酸に加えて,脂肪酸が肝臓において代謝されてできるケトン体がエネルギー源として全身で利用される.このような条件下では,肝臓におけるFGF21の産生が強く誘導されることなどから,ケトン体産生にFGF21が重要な役割を果たす可能性が示唆されていた29, 30).しかし,我々の検討では,ケトン食飼育により野生型マウスに比べFGF21ノックアウトマウスの白色脂肪組織におけるインスリン感受性が亢進していたものの,肝臓におけるケトン体合成に差は認められなかった46).一方,我々とは異なるグループからは,ケトン食飼育時の血中ケトン体濃度が,野生型マウスに比べFGF21ノックアウトマウスにおいて低いことが報告されている43).また,絶食に関しても,FGF21ノックアウトマウスの肝臓におけるケトン体合成や糖新生,脂肪酸酸化が抑制されたとの報告がある一方で,我々のノックアウトマウスでは,血中ケトン体濃度は減少せず,むしろ上昇していた42, 44).個々の解析において,マウスのバックグラウンドや,使用された餌の組成,細かい実験条件が異なるために,FGF21ノックアウトマウスの表現型が一致しないのかもしれない.一方,アミノ酸やタンパク質制限食負荷時における表現型についても解析が進められている.タンパク質含量を制限した食餌によりマウスを飼育した場合,エネルギー消費が亢進し体重が減少するが,FGF21ノックアウトマウスではこの変化が起こらない33).また,ロイシン制限食により起こる白色脂肪組織や肝臓におけるトリグリセリド含量の減少も,FGF21ノックアウトマウスでは認められなかった34).メチオニン/コリン制限食の負荷によりNASHモデル動物を作製することができるが,FGF21ノックアウトマウスを同条件で飼育すると肝臓における脂肪酸酸化が減少し,肝臓中のトリグリセリド含量が増加することが報告された35).一方で,FGF21の産生が誘導される肥満症に関しては,我々も含めFGF21ノックアウトマウスを用いた解析が行われているが,高脂肪食負荷による肥満症の発症や脂肪組織重量,肥満症に伴う糖脂質代謝に大きな差が生じることはなかった36).
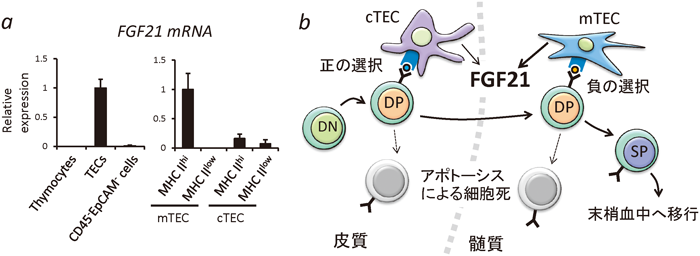
また,栄養摂取の変化以外のさまざまな状況下でのFGF21の生理的な意義も明らかにされつつある.たとえば,寒冷条件下で飼育したFGF21ノックアウトマウスでは白色脂肪組織におけるベージュ細胞の出現が著しく抑制される12).また,FGF21ノックアウトマウスでは,イソプロテレノール刺激による心肥大が亢進することや,虚血再還流により心筋梗塞を誘発した場合,梗塞部位が拡大することも明らかにされている41, 47).FGF21ノックアウトマウスに対し,膵炎を誘導した場合,野生型マウスに比較して膵臓の腺房細胞の破壊が有意に増加する40).また筋肉や肝臓において特異的にオートファジーを抑制したマウスにおいては,高脂肪食による肥満症の発症が抑制される.Kimらは我々のFGF21ノックアウトマウスを用い,この肥満症の抑制にFGF21が必須の役割を果たすことを明らかにした48).以上のように,FGF21は,摂取する栄養素の変化だけではなく,さまざまな状況の変化に対応する役割を持つことが明らかになりつつある.現在,我々も,代謝以外の役割の解明ということで,FGF21の発現組織の一つである胸腺における生理的意義を検討している.胸腺は,骨髄において産生される前駆T細胞の分化や成熟の場として知られている組織であり,解剖学的に皮質と髄質に分かれている.骨髄において産生された前駆T細胞は,血流を介して胸腺に流入した後,胸腺上皮細胞などとの相互作用を通じて,成熟したT細胞へと分化成熟し,再度血中へと放出される.図4aに記したように,FGF21は,T細胞系列ではなく,皮質,あるいは髄質の上皮細胞において特異的に発現していた.現在,FGF21ノックアウトマウス胸腺の細胞集団の詳細な検討や,胸腺器官培養系の解析を行っており,FGF21はT細胞の分化,成熟に関わる可能性が示唆されている(未公開データ)(図4b).
FGF21の薬理作用と,生理的意義に関して概説した.PubMedにおいて「FGF21」を検索すると,2000年のFGF21の単離同定から2005年の薬理作用の報告までの間は2報しかhitしないのに,現在では700報を超えており,その中には,紙面の都合上紹介できなかった重要な知見も数多く存在する.そして,今もFGF21に関する論文は増加しつつある.このFGF21研究の急激な発展には,新たな医療の開発を目指す研究者がFGF21研究に参入したことが大きく貢献していることは間違いない.しかし,内分泌因子であり,共受容体がKlothoで,形態形成因子というより代謝調節因子であるなど,従来のFGFからは予想できなかったFGF21の特徴が明らかにされ,基礎生物学的にも注目されたことも大きく貢献しているものと思われる.
では,今後のFGF21研究に残された課題はどんなものであろうか? まず,薬理作用に関しては,実験動物を用いた薬理作用のメカニズムの解明や,改変体の開発から一歩先に進んで,ヒトに対する投与を前提とした臨床的な研究が重要になると思われる.実際に一部の改変体に関しては,ヒトへの投与成績が報告され始めており,今後も増加していくことが期待される.また,内因性のFGF21産生を誘導するような薬品や食品などを発見することも,重要な試みになると思われる.一方で,生理的な意義に関する研究では,代謝に直接関わることが知られている脂肪組織や膵臓に加え,中枢神経系や心筋などにおいても,その機能維持に重要な役割を担うことが明らかになりつつある.FGF21は内分泌因子であるため,受容体と共受容体が存在する細胞であれば,産生部位との物理的な距離に関わらず,その機能の調節に関わる可能性が存在する.このような観点をもとに,FGF21の生理的な意義について検討を進めていけば,今後,たとえば絶食のような,FGF21の血中濃度が上昇するような栄養状態の変化と,特定の組織の機能維持との間に,予想もしえなかった因果関係が見つかるのではないかと期待している.
引用文献References
1) Itoh, N. (2010) Cell Tissue Res., 342, 1–11.
2) Nishimura, T., Nakatake, Y., Konishi, M., & Itoh, N. (2000) Biochim. Biophys. Acta, 1492, 203–206.
3) Goetz, R., Ohnishi, M., Kir, S., Kurosu, H., Wang, L., Pastor, J., Ma, J., Gai, W., Kuro-o, M., Razzaque, M.S., & Mohammadi, M. (2012) J. Biol. Chem., 287, 29134–29146.
4) Adams, A.C., Yang, C., Coskun, T., Cheng, C.C., Gimeno, R.E., Luo, Y., & Kharitonenkov, A. (2012) Mol. Metab., 2, 31–37.
5) Ding, X., Boney-Montoya, J., Owen, B.M., Bookout, A.L., Coate, K.C., Mangelsdorf, D.J., & Kliewer, S.A. (2012) Cell Metab., 16, 387–393.
6) Micanovic, R., Raches, D.W., Dunbar, J.D., Driver, D.A., Bina, H.A., Dickinson, C.D., & Kharitonenkov, A. (2009) J. Cell. Physiol., 219, 227–234.
7) Kharitonenkov, A., Shiyanova, T.L., Koester, A., Ford, A.M., Micanovic, R., Galbreath, E.J., Sandusky, G.E., Hammond, L.J., Moyers, J.S., Owens, R.A., Gromada, J., Brozinick, J.T., Hawkins, E.D., Wroblewski, V.J., Li, D.S., Mehrbod, F., Jaskunas, S.R., & Shanafelt, A.B. (2005) J. Clin. Invest., 115, 1627–1635.
8) Rydén, M. (2009) Cell. Mol. Life Sci., 66, 2067–2073.
9) Kharitonenkov, A., Wroblewski, V.J., Koester, A., Chen, Y.F., Clutinger, C.K., Tigno, X.T., Hansen, B.C., Shanafelt, A.B., & Etgen, G.J. (2007) Endocrinology, 148, 774–781.
10) Lin, Z., Tian, H., Lam, K.S., Lin, S., Hoo, R.C., Konishi, M., Itoh, N., Wang, Y., Bornstein, S.R., Xu, A., & Li, X. (2013) Cell Metab., 17, 779–789.
11) Joki, Y., Ohashi, K., Yuasa, D., Shibata, R., Ito, M., Matsuo, K., Kambara, T., Uemura, Y., Hayakawa, S., Hiramatsu-Ito, M., Kanemura, N., Ogawa, H., Daida, H., Murohara, T., & Ouchi, N. (2015) Biochem. Biophys. Res. Commun., 459, 124–130.
12) Fisher, F.M., Kleiner, S., Douris, N., Fox, E.C., Mepani, R.J., Verdeguer, F., Wu, J., Kharitonenkov, A., Flier, J.S., Maratos-Flier, E., & Spiegelman, B.M. (2012) Genes Dev., 26, 271–281.
13) Wente, W., Efanov, A.M., Brenner, M., Kharitonenkov, A., Köster, A., Sandusky, G.E., Sewing, S., Treinies, I., Zitzer, H., & Gromada, J. (2006) Diabetes, 55, 2470–2478.
14) Hughes, S.E. (1997) J. Histochem. Cytochem., 45, 1005–1019.
15) Zhang, Y., Lei, T., Huang, J.F., Wang, S.B., Zhou, L.L., Yang, Z.Q., & Chen, X.D. (2011) Mol. Cell. Endocrinol., 342, 41–47.
16) Lin, X., Li, G., He, X., Ma, X., Zhang, K., Zhang, H., Zeng, G., & Wang, Z. (2014) Mol. Cell. Biochem., 39, 33–42.
17) Hsuchou, H., Pan, W., & Kastin, A.J. (2007) Peptides, 28, 2382–2386.
18) Owen, B.M., Ding, X., Morgan, D.A., Coate, K.C., Bookout, A.L., Rahmouni, K., Kliewer, S.A., & Mangelsdorf, D.J. (2014) Cell Metab., 20, 670–677.
19) Douris, N., Stevanovic, D.M., Fisher, F.M., Cisu, T.I., Chee, M.J., Nguyen, N.L., Zarebidaki, E., Adams, A.C., Kharitonenkov, A., Flier, J.S., Bartness, T.J., & Maratos-Flier, E. (2015) Endocrinology, 156, 2470–2481.
20) Zhang, C., Shao, M., Yang, H., Chen, L., Yu, L., Cong, W., Tian, H., Zhang, F., Cheng, P., Jin, L., Tan, Y., Li, X., Cai, L., & Lu, X. (2013) PLoS ONE, 8, e82275.
21) Feingold, K.R., Grunfeld, C., Heuer, J.G., Gupta, A., Cramer, M., Zhang, T., Shigenaga, J.K., Patzek, S.M., Chan, Z.W., Moser, A., Bina, H., & Kharitonenkov, A. (2012) Endocrinology, 15, 2689–2700.
22) Yu, Y., Li, S., Liu, Y., Tian, G., Yuan, Q., Bai, F., Wang, W., Zhang, Z., Ren, G., Zhang, Y., & Li, D. (2015) Int. Immunopharmacol., 25, 74–82.
23) Planavila, A., Redondo-Angulo, I., Ribas, F., Garrabou, G., Casademont, J., Giralt, M., & Villarroya, F. (2015) Cardiovasc. Res., 106, 19–31.
24) Mu, J., Pinkstaff, J., Li, Z., Skidmore, L., Li, N., Myler, H., Dallas-Yang, Q., Putnam, A.M., Yao, J., Bussell, S., Wu, M., Norman, T.C., Rodriguez, C.G., Kimmel, B., Metzger, J.M., Manibusan, A., Lee, D., Zaller, D.M., Zhang, B.B., DiMarchi, R.D., Berger, J.P., & Axelrod, D.W. (2012) Diabetes, 61, 505–512.
25) Véniant, M.M., Komorowski, R., Chen, P., Stanislaus, S., Winters, K., Hager, T., Zhou, L., Wada, R., Hecht, R., & Xu, J. (2012) J. Endocrinol., 153, 4192–4203.
26) Kharitonenkov, A., Beals, J.M., Micanovic, R., Strifler, B.A., Rathnachalam, R., Wroblewski, V.J., Li, S., Koester, A., Ford, A.M., Coskun, T., Dunbar, J.D., Cheng, C.C., Frye, C.C., Bumol, T.F., & Moller, D.E. (2013) PLoS ONE, 8, e58575.
27) Gaich, G., Chien, J.Y., Fu, H., Glass, L.C., Deeg, M.A., Holland, W.L., Kharitonenkov, A., Bumol, T., Schilske, H.K., & Moller, D.E. (2013) Cell Metab., 18, 333–340.
28) Zhang, J. & Li, Y. (2014) Drug Discov. Today, 19, 579–589.
29) Badman, M.K., Pissios, P., Kennedy, A.R., Koukos, G., Flier, J.S., & Maratos-Flier, E. (2007) Cell Metab., 5, 426–437.
30) Inagaki, T., Dutchak, P., Zhao, G., Ding, X., Gautron, L., Parameswara, V., Li, Y., Goetz, R., Mohammadi, M., Esser, V., Elmquist, J.K., Gerard, R.D., Burgess, S.C., Hammer, R.E., Mangelsdorf, D.J., & Kliewer, S.A. (2007) Cell Metab., 5, 415–425.
31) Kim, H., Mendez, R., Zheng, Z., Chang, L., Cai, J., Zhang, R., & Zhang, K. (2014) Endocrinology, 155, 769–782.
32) Li, Y., Wong, K., Giles, A., Jiang, J., Lee, J.W., Adams, A.C., Kharitonenkov, A., Yang, Q., Gao, B., Guarente, L., & Zang, M. (2014) Gastroenterology, 146, 539–549.
33) Laeger, T., Henagan, T.M., Albarado, D.C., Redman, L.M., Bray, G.A., Noland, R.C., Münzberg, H., Hutson, S.M., Gettys, T.W., Schwartz, M.W., & Morrison, C.D. (2014) J. Clin. Invest., 124, 3913–3922.
34) De Sousa-Coelho, A.L., Relat, J., Hondares, E., Pérez-Martí, A., Ribas, F., Villarroya, F., Marrero, P.F., & Haro, D. (2013) J. Lipid Res., 54, 1786–1797.
35) Fisher, F.M., Chui, P.C., Nasser, I.A., Popov, Y., Cunniff, J.C., Lundasen, T., Kharitonenkov, A., Schuppan, D., Flier, J.S., & Maratos-Flier, E. (2014) Gastroenterology, 147, 1073–1083.
36) Fisher, F.M., Chui, P.C., Antonellis, P.J., Bina, H.A., Kharitonenkov, A., Flier, J.S., & Maratos-Flier, E. (2010) Diabetes, 59, 2781–2789.
37) Dushay, J.R., Toschi, E., Mitten, E.K., Fisher, F.M., Herman, M.A., & Maratos-Flier, E. (2014) Mol. Metab, 4, 51–57.
38) Hondares, E., Iglesias, R., Giralt, A., Gonzalez, F.J., Giralt, M., Mampel, T., & Villarroya, F. (2011) J. Biol. Chem., 286, 12983–12990.
39) Kim, K.H., Kim, S.H., Min, Y.K., Yang, H.M., Lee, J.B., & Lee, M.S. (2013) PLoS ONE, 8, e63517.
40) Johnson, C.L., Weston, J.Y., Chadi, S.A., Fazio, E.N., Huff, M.W., Kharitonenkov, A., Köester, A., & Pin, C.L. (2009) Gastroenterology, 137, 1795–1804.
41) Planavila, A., Redondo, I., Hondares, E., Vinciguerra, M., Munts, C., Iglesias, R., Gabrielli, L.A., Sitges, M., Giralt, M., van Bilsen, M., & Villarroya, F. (2013) Nat. Commun., 4, 2019.
42) Hotta, Y., Nakamura, H., Konishi, M., Murata, Y., Takagi, H., Matsumura, S., Inoue, K., Fushiki, T., & Itoh, N. (2009) Endocrinology, 150, 4625–4633.
43) Badman, M.K., Koester, A., Flier, J.S., Kharitonenkov, A., & Maratos-Flier, E. (2009) Endocrinology, 150, 4931–4940.
44) Potthoff, M.J., Inagaki, T., Satapati, S., Ding, X., He, T., Goetz, R., Mohammadi, M., Finck, B.N., Mangelsdorf, D.J., Kliewer, S.A., & Burgess, S.C. (2009) Proc. Natl. Acad. Sci. USA, 106, 10853–10858.
45) Dutchak, P.A., Katafuchi, T., Bookout, A.L., Choi, J.H., Yu, R.T., Mangelsdorf, D.J., & Kliewer, S.A. (2012) Cell, 148, 556–567.
46) Murata, Y., Nishio, K., Mochiyama, T., Konishi, M., Shimada, M., Ohta, H., & Itoh, N. (2013) PLoS ONE, 8, e69330.
47) Liu, S.Q., Roberts, D., Kharitonenkov, A., Zhang, B., Hanson, S.M., Li, Y.C., Zhang, L.Q., & Wu, Y.H. (2013) Sci. Rep., 3, 2767.
48) Kim, K.H., Jeong, Y.T., Oh, H., Kim, S.H., Cho, J.M., Kim, Y.N., Kim, S.S., Kim, H., Hur, K.Y., Kim, H.K., Ko, T., Han, J., Kim, H.L., Kim, J., Back, S.H., Komatsu, M., Chen, H., Chan, D.C., Konishi, M., Itoh, N., Choi, C.S., & Lee, M.S. (2013) Nat. Med., 19, 83–92.