哺乳動物の神経系の形成において,神経細胞は非常に長い距離(自分自身の大きさの数十倍以上)を移動し最終的な場所にたどり着く.その結果,機能の類似した神経細胞は「層」または「神経核」と呼ばれる構造を形成する.これらの構造は,軸索誘導や樹状突起発達を効率的または正確にし,神経回路網が正常に形成・維持されるために必要であると考えられる.言い換えれば,神経細胞移動の異常は神経回路網の形成や機能の異常に直結し,何らかの病態につながる可能性が高いことが容易に想像される.しかし神経細胞移動は神経細胞分化とも密接に関与する複雑な現象であり,かつてはそのアプローチ法も限定されていたため,多くの「モデル」や「仮説」が提唱されていたもののその是非は不明であった.1990年代以降,神経発生に関わる遺伝子・分子が多く同定されるとともに,イメージング技術の進歩や疾患遺伝子の同定などもあいまって,神経細胞移動の分子機構とその異常によって生じる疾患の理解は急速に進んだ.
神経細胞が目的位置までたどり着き,止まるためには,「細胞外」「細胞内」両方での制御が必要である.前者には,細胞を誘因あるいは反発する分子や接着分子が含まれる.特に,分泌タンパク質リーリンは,その欠損により層構造を持つ神経系のほぼすべてが異常になるという点で,機能解明に最も注目が集まっている分子である.一方,後者には,細胞骨格系とこれを制御する分子や遺伝子発現変動に関わる分子が含まれる.本稿では,前者に焦点をあて,リーリンの機能制御機構,および神経細胞移動における機能を論じる.後者については他の総説を参照いただきたい.
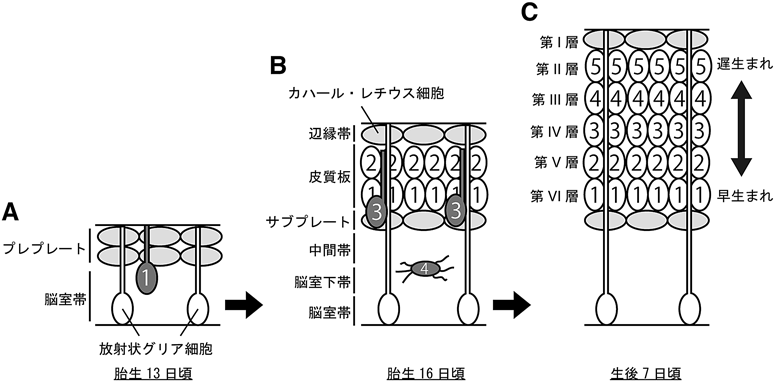
哺乳類の大脳皮質は類似した形態や機能を持つ神経細胞が六つに分類される層を形成する.大脳皮質を構成する神経細胞には,興奮性神経細胞と抑制性神経細胞の2種類があり,それぞれ誕生する場所や移動様式が異なる.大脳皮質の神経細胞の約8割を占める興奮性神経細胞は,脳室帯に存在する神経幹細胞である放射状グリア細胞から分化して生じ,脳室側から表層側へと放射状に移動する[放射状移動(radial migration),図1].その際,後から移動してきた(遅生まれの)神経細胞はすでに移動を終えた(早生まれの)神経細胞を追い越し表層側へ移動する.そのため,遅生まれの神経細胞ほど表層側の層に配置し,早生まれの神経細胞は脳室側に配置される(図1).また,誕生日が近い神経細胞どうしが一つの層を形成する.このような構造はinside-out型層構造と呼ばれ,神経細胞間のネットワーク形成を容易にするために重要である1).また,抑制性神経細胞は大脳基底核原基の神経幹細胞から生まれ,大脳皮質内を接線方向に移動する[接線移動(tangential migration)].抑制性神経細胞は生まれる場所の違いによりいくつかのサブタイプに分類され,それぞれ大脳皮質へ向かう際の経路が異なることが知られている2).しかし,最終的な大脳皮質での配置は,おおむねinside-out様式で配置されることがわかっている3).抑制性神経細胞の移動は興奮性神経細胞の移動に比べ長く,いくつかの細胞遊走因子,誘引因子や反発因子などにより制御されている.その制御機構は,抑制性神経細胞の起源や誕生日により異なり,興奮性神経細胞のそれとは共通しない部分も多い.詳細は他の総説4)を参考にしていただきたい.本稿では紙面の都合上,興奮性神経細胞の移動機構に焦点をあて紹介する.
大脳皮質形成の初期段階では,最初に生まれた神経細胞がプレプレートを形成する(図1).その後,脳室帯で生まれた神経細胞がプレプレートを分け入って侵入し,プレプレートは辺縁帯とサブプレートの二つに分かれる(プレプレートスプリッティング).プレプレートスプリッティングが起きた後,神経細胞はサブプレートを越えて移動し,先に述べたように早生まれの神経細胞を追い越し,辺縁帯の直前で移動を終える.これを繰り返し行いinside-outのパターンで皮質板が形成される(図1).胎生期の大脳皮質神経細胞の移動は,大雑把にはこのように説明されてきたが,近年,神経細胞のより高度な標識方法が開発されたことにより,神経細胞がいくつかの異なった移動様式をとることが明らかになってきた.
興奮性神経細胞の移動様式は,ロコモーション(locomotion),細胞体トランスロケーション(somal translocation),多極性移動(multipolar migration)の三つに分類される.ロコモーションは,放射状グリア細胞の放射状線維を足場にして神経細胞が移動する様式であり,神経細胞は先導突起を持ち,先導突起の伸長と細胞体の引き上げを繰り返して表層側に向かって移動する(図1B)5).一方,細胞体トランスロケーションでは,神経細胞は放射状線維を足場とせずに移動する.この様式では,神経細胞は自身の突起を表層まで長く伸ばし,突起を短くしながら細胞体を移動する(図1A)6).多極性移動は,神経細胞が多極性の形態をとり移動する様式であり,脳室下帯付近の神経細胞によくみられる.この様式では神経細胞は多くの突起を伸ばしたり縮めたりを繰り返し,ゆっくりと表層側に移動する(図1B)7).神経細胞の生まれる時期によって用いる移動様式が異なることが知られており,早生まれ[胎生12日(マウスの場合,以下同じ)生まれ]の神経細胞は,細胞体トランスロケーションの様式で皮質板に侵入する6)(図1A).一方,遅生まれ(胎生14日生まれ)の神経細胞は,多極性移動を経て皮質板に侵入したのち,ロコモーション様式により表層付近まで移動する(図1B).その後,先導突起が辺縁帯に届くと放射状グリア線維から細胞体が離れ,先導突起を短くするように表層側に細胞体を引き上げ移動が完了する.この様式は,ターミナルトランスロケーション様式と呼ばれる6, 8).このように,大脳皮質の興奮性神経細胞は,いくつもの移動様式を用いて長い距離を移動し,最終的な位置に配置される(図1C).
胎生期の神経細胞移動異常は先天性の脳発達障害の原因であり,その代表例としては滑脳症があげられる.滑脳症の患者の脳には脳回(すなわち「しわ」)がなく,精神遅滞,てんかん発作や筋痙縮などの症状を示す.滑脳症の原因としては,Lis1, doublecortin, tubulin alpha 1AそしてReelinなどの遺伝子変異が報告されており,原因遺伝子の違いにより症状の重篤さが異なる9).滑脳症の中でも最も多く症例が報告されているのがMiller-Dieker症候群である.この患者では脳の全体で脳回がまったく存在せず,滑脳症の中でも重篤な症状を呈する.Miller-Dieker症候群の原因は,17番染色体にあるLis1(PAFAH1B1)遺伝子の欠損であり10, 11),17番染色体短腕部分での微小欠失や転座が報告されている12, 13).Lis1タンパク質は微小管モータータンパク質である細胞質ダイニンと結合し,核移動,タンパク質の軸索輸送を制御する14).また,Lis1ノックアウトマウスでは,神経細胞の移動が遅く大脳皮質が正常にできない15).したがって,Lis1は細胞骨格を制御することで,神経細胞の適切な移動を制御することがわかる.Lis1をはじめ滑脳症の原因遺伝子産物の多くは細胞内タンパク質であり,細胞内で細胞の形を制御する役割を持つ.一方で,滑脳症原因遺伝子の産物のうちリーリンは唯一の細胞外因子である16).
リーリンについての研究は,約60年以上前に発見された,ある自然発症マウスに端を発する.1951年に運動失調を呈する自然発症マウスが発見され,このマウスは千鳥足のような歩き方(reeling gait)をするため,リーラー(reeler)マウスと名づけられた17).リーラーマウスには多くの脳構造異常が認められる.たとえば,運動をつかさどる小脳では層構造が正常に形成されず,サイズも非常に小さくなる.また,海馬のアンモン角および歯状回の神経細胞は散在し,大脳皮質の層構造はおおむね逆転する18).そのため,リーラーマウスの原因遺伝子産物は神経細胞の配置,すなわち正常な神経細胞の移動に必須な分子であることが推察された.
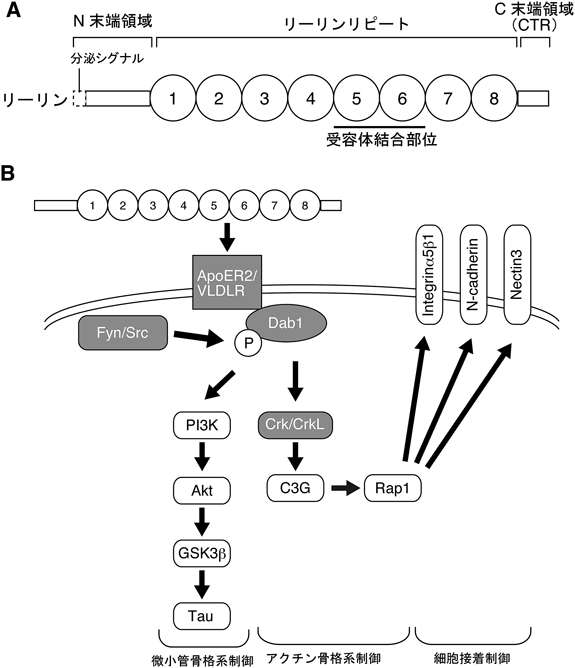
1995年にリーラーの原因遺伝子の全長cDNAが同定され,リーリン(Reelin)と名づけられた19).リーリンは,神経発生に関与する他のタンパク質と比べ非常に巨大な分子(マウスでは全長3461アミノ酸残基からなる)であり,糖鎖修飾を受けその大きさは400 kDaを超える20).リーリンは,分泌シグナルを含むN末端領域,8回の繰り返し構造(リーリンリピート),そして塩基性アミノ酸に富むC末端領域(CTR)からなる19)(図2A).リーリンと相同性の高いタンパク質はショウジョウバエや線虫にはなく,また,高等動物でもファミリー分子は存在しない.これらの事実は,神経発生に重要な多くの細胞外分子(Wnt, Semaphorin, Netrin, Slitなど)が進化の過程で高度に保存され,かつ高等動物ではファミリーを形成していることと対照的である.
リーリンは,胎生期には大脳皮質や,海馬の辺縁帯に位置するカハール・レチウス細胞や,小脳の顆粒細胞に主に発現し,生後の大脳皮質では抑制性神経細胞に発現する21–24).リーリンは,これらリーリン産生細胞から分泌され細胞外を拡散し,移動中の神経細胞に作用すると考えられる.しかし,リーリンの細胞外での挙動を可視化することは難しく,リーリン産生細胞からリーリンがどの程度拡散できるのかは明確にはわかっていない.胎生期の大脳皮質では,移動前の神経細胞や,脳室帯の神経幹細胞にリーリン受容体が発現すること25)や,脳室帯での神経新生をリーリンが制御する報告26)があることを考えると,リーリンはある程度遠くまで拡散して作用していると思われる.
リーリンは,リーリンリピートの5番目,6番目を介し27),神経細胞膜上に発現するアポリポタンパク質E受容体2(ApoER2),もしくは超低比重リポタンパク質受容体(VLDLR)に結合する28, 29).リーリンがこれら受容体に結合すると,FynなどのSrcファミリーキナーゼが活性化し30),細胞内タンパク質Dab1のチロシンリン酸化が誘導される31)(図2B).ApoER2とVLDLRをともに欠くマウス32),Dab1欠損マウス33, 34),Dab1のチロシン残基に変異を導入したマウス35),SrcファミリーキナーゼのうちFynとSrcをともに欠くマウス36)はいずれもリーラーマウスと酷似した大脳皮質形成異常を示す.したがって,ApoER2/VLDLRを介したリーリンによるDab1のチロシンリン酸化が正常な大脳皮質形成に必要であると考えられる.
Dab1はTyr 185, Tyr 198, Tyr 200, Tyr 220, Tyr 232の5か所のチロシンリン酸化部位を持ち35),リーリンの刺激によりリン酸化を受ける.Tyr 185とTyr 198でリン酸化を受けたDab1はPI3Kのp85αサブユニットに結合し37),Aktのリン酸化およびGSK3βのリン酸化を誘導する.これによりTauのリン酸化が制御される29, 38)(図2B).また,Dab1のTyr 220およびTyr 232のリン酸化は,Crk/CrkL-C3G複合体をリクルートし,低分子量Gタンパク質であるRap1の活性化を促す39–41).また,リン酸化されたDab1は速やかに分解される31).この分解にはE3ユビキチンリガーゼのCullin5が必要であり,これを移動中の神経細胞においてノックダウンすると,神経細胞は適切な場所で移動を停止することができず,表層近くまで移動してしまう42)すなわち,リン酸化Dab1の分解は,神経細胞の移動停止に必要であることが示唆される.Dab1の下流分子としては他にも数多くの候補分子があげられているが,その多くは微小管・アクチン細胞骨格系や細胞接着を制御する働きを持ち,リーリンがこれら分子の機能を調節し,正常な神経細胞移動を制御すると予想される(図2B).どの分子が神経細胞移動のどのステップで重要であるのかの全貌はいまだ明らかになっていないが,後述するようにその一部が解明されつつある.
受容体結合に必要なリーリンの最小領域は,5番目と6番目の二つのリーリンリピートであるが,この二つのリピートだけでは下流シグナルを十分に活性化することはできない27).
また,リーリンのN末端領域,もしくはC末端領域を欠くリーリンは,Dab1のリン酸化誘導能が著しく低下する43, 44).N末端領域がリーリンの二量体化に必要であること43)から,二量体化したリーリンがリーリン受容体をクラスタリングし,効率的なシグナル伝達を行うモデルが提唱された45).また,C末端領域はリーリンの多量体化には寄与しないが,リーリン受容体へのリーリンの親和性を上げることがわかり,C末端領域が未知共受容体に結合し効率的なシグナル伝達を行うモデルが示された44, 46).以上から,リーリンのN末端領域とC末端領域は,効率的なリーリンの下流シグナル活性能に必要であると考えられる.
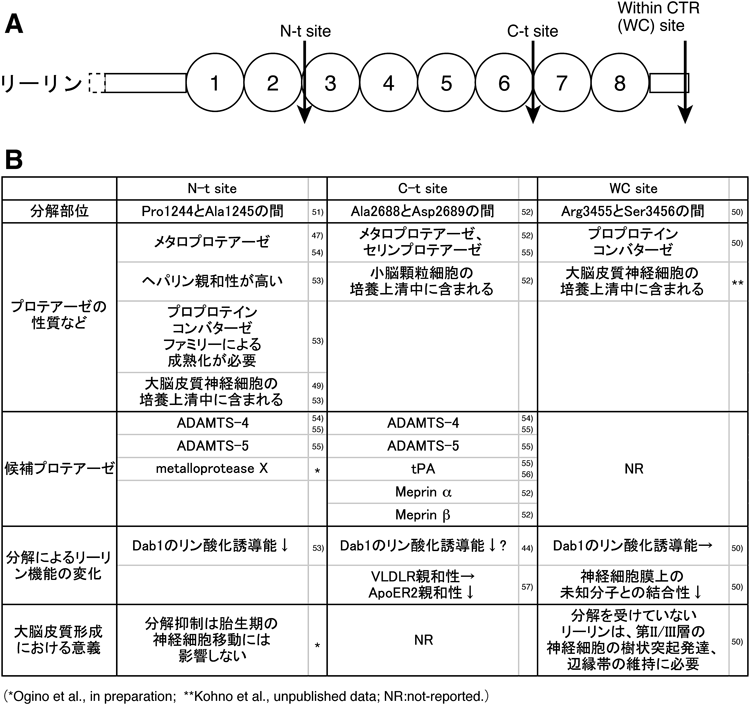
細胞外に分泌されたリーリンは,3か所で分解(プロテオリシス)を受ける47–50)(図3A).我々は,リーリンの分解部位をアミノ酸レベルで同定し,リーリンリピート3の内部(N-t site51)),リーリンリピートの6と7の間(C-t site52)),そしてCTRの内部(within CTR site:WC site50))でプロテアーゼによる特異的な分解を受けることを明らかにした.プロテアーゼによる分解は,多くの分泌タンパク質や膜タンパク質の機能を制御する.そのため,リーリンの機能もプロテオリシスにより制御され,大脳皮質形成にも重要な役割を果たす可能性が考えられる.しかし,分解を担うプロテアーゼはいまだ同定されておらず,生体内におけるリーリン分解の意義は,ほとんどわかっていない.我々は,この点を明らかにすることを目的に研究を行い,少しずつだがリーリンのプロテオリシスによる機能制御機構とその意義がわかってきた.次に分解部位ごとにわかっている知見をまとめてみたい(図3B).
1)N-t siteにおけるリーリン分解
リーリン分解を担う酵素については,以下のことが報告されている.①大脳皮質培養神経細胞の培養上清に含まれること49),②強いN-t site分解活性を持つこと49, 53),③2価金属イオンを必要とするメタロプロテアーゼの一種であること47),④活性発現にはプロプロテインコンバターゼ(PC)ファミリーによる成熟化が必要であること53),⑤ヘパリンへ強い親和性を持つこと53),である.我々は,このプロテアーゼを部分精製しその性状を解析した結果,リーリンがN-t siteで分解を受けると,Dab1リン酸化誘導能が著しく低下することを見いだした53).すなわち,N-t siteでのリーリン分解はリーリンの生理活性を負に制御する役割を持つ.
我々を含むいくつかの研究グループが,リーリン分解を担うプロテアーゼの同定を試み,いくつかの候補があげられた54, 55).しかし,これら候補分子が実際に生体内でのリーリン分解に関与しているかについては不明である.我々は最近,大脳皮質神経細胞培養上清からとある分泌型プロテアーゼをN-t site分解プロテアーゼとして同定することに成功した(Oginoら,投稿準備中).このプロテアーゼは,先に述べた性質①~⑤をすべて満たしており,また,そのノックアウトマウスの大脳皮質では,N-t siteでのリーリン分解が抑制されていた.さらに,Dab1の量が減少していることから,リーリンのシグナル活性が増強されていることが強く示唆された.興味深いことに,このノックアウトマウスでは胎生期の大脳皮質形成は正常であった(Oginoら,投稿準備中).そのため,少なくとも胎生期の大脳皮質形成にはリーリンのN-t siteでの分解は必須ではなく,過剰のリーリンシグナルが層構造形成に悪影響を与えることはないことが示唆される.
2)C-t siteにおけるリーリン分解
N-t site分解活性を持つプロテアーゼとして報告されたADAMTS-4,-5はC-t site分解活性も合わせ持つ54, 55).また,薬剤誘導性の長期増強を引き起こした際にC-t site分解が増加し,このときのリーリン分解に組織プラスミノーゲンアクチベーター(tPA)が関与することが報告された56).しかし,無刺激のtPAノックアウトマウスでは,リーリン分解が抑制されなかったため,通常はtPA以外のプロテアーゼがリーリン分解を担うことが示唆された56).我々は,C-t siteでのリーリン分解活性は,大脳皮質神経細胞の培養上清にはほとんど含まれないが,小脳顆粒細胞の培養上清にはN-t site分解活性に加え,C-t site分解活性も含まれることを見いだした52).C-t site周辺の配列から,meprin αとmerin βがリーリン分解プロテアーゼとして候補にあがり,実際これらはリーリン分解活性を持つことがわかった52).しかし,meprin βノックアウトマウスの大脳,小脳では,リーリン分解が抑制されなかったため,生体内でのリーリン分解への寄与は低いと思われる52).
C-t site分解によるリーリン機能制御については,いまだ明らかになっていない.しかし,N-t siteとC-t siteの両方で分解を受けて生じる中央断片(RR36)はN-t siteだけで分解を受けた結果生じる断片(RR38C)に比べてApoER2への結合能が低いこと57),およびCTRがリーリンの下流シグナル伝達能に必要であること44)から,C-t siteでのリーリン分解はN-t siteでの分解と同様にリーリンの下流シグナル伝達能を負に制御することが推察される.また興味深いことに,VLDLRに対する結合能はRR36とRR38Cの間で有為な差が認められない57)ため,C-t siteにおける分解は,リーリンのApoER2とVLDLRに対する結合性を調節し,個別の機能発現に関与する可能性が考えられる.
3)第三の分解部位WC siteにおけるリーリン分解
我々は,リーリンがCTRの内部でPCファミリーのプロテアーゼにより分解を受け,CTRのC末端から6アミノ酸残基のみが遊離することを新たに見いだした50).すなわち,完全長CTRを持つリーリン(リーリンFL)から,C末端から6残基を欠くリーリン(リーリンΔ6)が生じる.この分解はN-t site分解,C-t site分解に次ぐ,第三のリーリン分解でありwithin CTR(WC)site分解と名づけた50).リーリンのN-t site, C-t siteの分解は複数のグループにより報告されたが,WC siteを見いだしたのは我々のグループだけであった.その理由は,リーリンFLとリーリンΔ6の分子量の差が非常に小さく,ウェスタンブロッティングでは分離することができないためであると考えている.我々は,CTRのC末端側にエピトープタグを挿入した変異体リーリンの発現プラスミドを作製し,これを培養細胞に発現した際に培養上清中のリーリン抗タグ抗体での検出を試みたことが,C末端側の6残基が欠失していることを発見したきっかけとなった.WC siteでのリーリン分解は,少なくともin vitroではリーリンのDab1リン酸化誘導能に影響しないことがわかった.しかし興味深いことに,リーリンΔ6に比べ,リーリンFLは神経細胞の膜に強く結合することを見いだした.このことは,リーリンと未知の分子との結合がWC site分解により制御されることを示している.WC site分解の大脳皮質形成における意義を考えるにあたって,我々はCTRを欠くリーリンを発現するノックインマウス(ΔC-KI)を作製し,その大脳皮質における表現型に着目した.ΔC-KIの大脳皮質層構造は,胎生期には正常に形成されるが,生後3日目以降に第II/III層の神経細胞のみが散在し辺縁帯に侵入すること,また第II/III層の神経細胞の樹状突起発達が悪いことがわかった.これらの知見は,CTRを介したリーリンシグナル伝達が辺縁帯とその直下の構造維持に必要であることを示す.そこで,WC site分解の辺縁帯形成における意義を調べるために,我々はリーリンの異所性発現系58)を用いた.この系でリーリンを脳室帯に異所性に発現すると,脳室付近に神経細胞からなる凝集塊ができ,リーリンが高濃度に存在する中央部には神経細胞が侵入しないことがわかった58).また,この凝集塊の構造は,大脳皮質の辺縁帯とその直下の皮質板の構造と酷似しており,辺縁帯付近でのリーリン機能を再現するよい実験モデルである58, 59).リーリンΔ6を異所性に発現した場合も,リーリンFLを発現した際と同様に脳室付近に神経細胞からなる凝集塊が形成された.しかし,リーリンFLに比べ,リーリンΔ6の発現によりできた凝集塊は中央の空洞部分の面積が小さく,空洞周辺の細胞密度が低いこと,すなわち空洞部分に神経細胞が侵入することがわかった50).このことから,辺縁帯様の構造を形成するためにはリーリンΔ6では不十分であり,リーリンFLが必要であることがわかった.以上の知見から,第II/III層の神経細胞の樹状突起発達と配置維持には,リーリンFLと未知の分子との結合が必要であり,この機能はPCファミリーによるプロテオリシスにより制御されることが示唆された50).リーリンFLに結合する分子は,リーリン受容体の共受容体として働き,生後の大脳皮質形成に重要な役割を持つ可能性がある.リーリンによる大脳皮質形成機構の詳細を明らかにするためには,この結合分子の同定が不可欠である.
リーラーマウスの大脳皮質形成異常から,リーリンが神経細胞移動に必須な役割を担うことは明白である.しかし,リーラーマウスの表現型のすべてがリーリンの機能を反映するとは限らない.たとえば,リーラーマウスでは樹状突起からなる異常な構造物が形成され,これに沿って神経細胞は移動を停止させる傾向がある60).この知見は,リーラーマウスの表現型の少なくとも一部は,異常構造物による物理的な障害であることを示唆する.したがって,単純にリーラーマウスの大脳皮質を解析しても直接的なリーリンの機能の欠損ではなく,二次的な影響をみてしまう可能性がある.近年,リーリン下流シグナル分子やリーリン受容体に対しての,RNA干渉法を用いたノックダウン実験や,条件的ノックアウトマウスを用いた実験(すなわち,リーラーマウスのように発生初期からのリーリンシグナル欠損ではなく,特定の時期における特定の神経細胞でのリーリンシグナル欠損)が精力的に行われ,これにより神経細胞移動におけるリーリンシグナルの意義が少しずつだがわかってきた.
1)神経細胞移動におけるリーリン下流シグナル分子の機能
リーリン下流シグナル因子の中で,最も上流で働く分子はDab1であり,大脳皮質形成におけるDab1の機能がこれまでに最も詳細に解析されている.遅生まれ(胎生14.5日目)の神経細胞において,Dab1をノックダウンもしくはノックアウトすると,神経細胞はロコモーション移動を正常に行い,表層近くまで移動する.しかし,神経細胞移動の最終ステップであるターミナルトランスロケーション様式での移動が異常になり,神経細胞は通常よりやや脳室側に配置する61, 62).このDab1依存的なターミナルトランスロケーションが開始される場所は,辺縁帯直下の未成熟神経細胞が多く存在する領域(原皮質帯)であり,Dab1は原皮質帯への進入に必要であること,またDab1依存的に原皮質帯内に侵入することが原皮質帯内でのinside-out様式での神経細胞配置に必要であることがわかった8).さらに,神経細胞が原皮質帯へ進入する際には,ApoER2/VLDLR-Dab1-Crk/CrkL-C3G-Rap1経路を介したIntegrinα5β1の活性化が必要であることが明らかになった(図2B参照)59).また,Rap1の活性化は,遅生まれの神経細胞が多極性移動からロコモーションへ移動様式を切り替える際にも必要であり,その下流でN-cadherinの機能が調節されることが報告された63).しかし,Dab1の条件的ノックアウトでは,多極性移動からロコモーションへの切り変わりは正常であるという報告がある62)ため,多極性移動におけるリーリン-Dab1-Rap1シグナルの重要性についてはさらなる検討が必要である.
一方,早生まれ(胎生12.5日目)の神経細胞におけるDab1の重要性は,Dab1条件的ノックアウトマウスを用いた研究により検討され,早生まれの神経細胞でDab1をノックアウトすると,細胞体トランスロケーションが異常になる62).この際においてもRap1の下流でN-cadherinの機能が調節されることが必要であることがわかった62).また,リーリンに依存したRap1の活性化がNectin3を介したN-cadherinの細胞膜上へのリクルートに必要であることが示され,ターミナルトランスロケーション中の神経細胞と辺縁帯のカハール・レチウス細胞間での,Nectin3–Nectin1のヘテロフィリック結合,およびN-cadherinのホモフィリックな結合が,正常な細胞体トランスロケーションに必要であることがわかった64).
また,上記のDab1のloss-of-function実験では樹状突起の発達に障害が起こる8, 61, 62).樹状突起異常は移動障害による影響である可能性が考えられるが,リーラーやDab1ノックアウトマウス由来の培養神経細胞を用いた研究から,リーリン-Dab1シグナルが神経細胞の樹状突起形成に必要であることが明らかになっている65, 66).そのため,Dab1のloss-of-functionによる樹状突起形成異常は移動障害の二次的な影響の結果ではないと思われる.むしろ,樹状突起の正常な発達が,神経細胞の配置に重要である報告もある67).
2)神経細胞移動におけるリーリン受容体(ApoER2, VLDLR)の機能
大脳皮質におけるApoER2とVLDLRの発現パターンは異なり,ApoER2は脳室帯付近に,VLDLRは皮質板に主に発現する25, 68).これら二つのリーリン受容体の大脳皮質形成における重要性がApoER2, VLDLRそれぞれの単独ノックアウトマウスを用いて検討された69).VLDLRノックアウトマウスでは,おおむね正常に層構造が形成されるが,第II/III層の神経細胞が辺縁帯に侵入する.そのため,VLDLRは遅生まれの神経細胞が辺縁帯の直下で停止するために必要であると考えられる.一方,ApoER2ノックアウトマウスでは,特に遅生まれの神経細胞の移動が異常となり,表層側まで移動できない.このことから,ApoER2はロコモーション移動する際に重要な役割を持つことが示唆される.
3)神経細胞移動におけるリーリンの機能
ここまでで,リーリンが大脳皮質における正常な神経細胞層の形成に必要であることは理解できると思うが,移動中の神経細胞に対してリーリンは一体何を行っているのであろうか? リーリンが同定され,主に辺縁帯のカハール・レチウス細胞に発現することがわかったころから,リーリンには誘引作用や反発作用があるのではないかと考えられてきた70).すなわち,リーリンは①脳室帯で生まれた神経細胞の移動をリーリン濃度の高い方に誘引する作用を持つ可能性と,②リーリン濃度の高い辺縁層に神経細胞が侵入しないように反発作用を持つ可能性である.しかし,これらの可能性を直接支持する証拠はいまだない.
また,リーリンが「停止シグナル」と「許容シグナル」を発揮する可能性がいくつかの研究結果から議論されてきた.停止シグナルはリーリンが移動中の神経細胞に作用すると移動を停止させる効果を示し,許容シグナルはリーリンには積極的な役割がなく,他の分子の補助的な役割を持ち,神経細胞の移動を許可するというものである.停止シグナル説は,2000年のAntonらの報告がきっかけとなり提唱された説である.Antonのグループは,リーリンがIntegrinα3β1と結合し,これにより神経細胞が放射状線維から離脱し,移動を停止することを示した71).また,Dab1のリン酸化がIntegrinα3の発現量を制御し,これにより神経細胞と放射状線維との接着性が調節され,放射状線維からの離脱が制御されることが報告された72).これらの知見から,リーリンはIntegrinα3β1との結合を介して,神経細胞の放射状線維からの離脱および移動の停止が制御されることが示唆された.しかし,移動中の神経細胞においてIntegrinβ1を欠失した条件的ノックアウトマウスや,Integrinα3ノックアウトマウスでは,リーラーのような層構造形成異常はみられなかった73)ため,現在は,Integrinα3β1を介したリーリンによる神経細胞停止機構については否定的な見解を持つ研究者が多い.
一方,許容シグナル説はCurranらの研究74)がきっかけとなり提唱された説である.彼らは表層側からのリーリン分泌が大脳皮質形成に必要かどうかを検討するために,Nestinプロモーター下でリーリンを発現するトランスジェニックマウス(このマウスでは,通常リーリンがほぼ存在しない脳室帯でリーリンが異所的に発現する)を作製した.このトランスジェニックマウスでは神経細胞の移動が阻害されず,大脳皮質の層構造は正常であった.さらに,このマウスをリーラーバックグラウンドにした場合(すなわち,内在性リーリンは存在せず,トランスジェニック由来のリーリンが脳室に発現する),リーラーのプレプレートスプリッティング異常が回復した.リーリンが脳室に異所性発現しても神経細胞移動が停止しないこと,また異所性の発現でもリーラーマウスの表現型が一部回復することから,リーリンは単純な停止シグナルとして働くのではなく,許容シグナル(自分自身はガイダンス機能を持たないが,他の分子が機能を発揮するための前提として必要)として働く可能性が考えられた.
Curranらの報告以外に許容シグナル説を支持する報告がいくつかある.たとえば,cortical hemと呼ばれる部位由来のカハール・レチウス細胞を遺伝学的手法により除去したマウス75)や,p73ノックアウト(辺縁層におけるカハール・レチウス細胞が激減する)マウス76)では,辺縁層付近におけるリーリン量が激減しているにも関わらず,プレプレートスプリッティングが起こり,層構造はおおむね正常に形成された.さらに,リーラーマウス由来の大脳皮質スライス培養系に,全長リーリンやリーリン中央部分断片を添加する(すなわち,リーリンの濃度勾配はない状態)実験でも,プレプレートスプリッティング異常が回復する48).以上の知見は,大脳皮質形成におけるリーリンの機能(少なくとも,プレプレートスプリッティングなどの一部の機能)には,リーリンが必ずしも辺縁帯のカハール・レチウス細胞から分泌される必要がないことを示唆する.そのため,リーリンは,神経細胞のリーリン以外の他の分子に対する反応性を変える許容シグナルとして考えられるようになった.
本稿では,大脳皮質における神経細胞移動と,その制御機構を巨大分泌タンパク質リーリンに着目し概説したが,結局リーリンは脳の中で何をしているの?という問いに対しては,一言では答えることができない.おそらく,リーリンには複数の機能があり,その機能を発揮するタイミングやターゲットがいくつも存在するのであろう.近年,神経細胞移動に必要な細胞内分子が同定され,各移動様式における重要性が少しずつだが明らかになってきた.しかし,これら分子の機能制御にリーリンがいつ,どのように必要であるのかは不明である.これを明らかにするためには,リーリンの生化学的性質,およびリーリンシグナルの機能制御機構を明らかにすることと,さらにそれらの神経細胞移動における意義を明らかにすることが必須である.しかし,リーリン機能の分子機構はなおざりにされ,リーリン下流分子の機能解析に焦点があたっているのが現状である.大脳皮質の神経細胞移動におけるリーリンの機能を明らかにする上で最も大きな妨げとなる理由として,リーリンが非常に大きい分子であることがあげられる.リーリンが大きな分子(エキソンが65個)であることは,リーリンの遺伝子改変マウスの作製を困難にし,我々がCTR欠損のノックインマウス50)を報告するまで成功例は皆無であった.最近,Herzらが,リーリンの条件的ノックアウトマウスの作製に成功した77).このマウスを利用し,胎生期の神経細胞移動に,いつからリーリンが必要であるかという疑問が近いうちに明らかになると期待される.また,リーリンは少なくとも3か所で分解を受ける.この分解がいつ,どこで,どれくらい起こるのか,また神経細胞移動への影響はどれくらいあるのかを詳細に明らかにするためには,リーリン分解酵素の同定,分解酵素のノックアウトマウスやトランスジェニックマウス,さらに分解抵抗性リーリンのノックインマウスの作製が必要となる.我々は現在,分解によるリーリンの機能制御とその意義の解明を精力的に進めており,これが明らかになればリーリンの真の機能の解明に微力ながら貢献できると考えている.今後,リーリンの機能制御機構が明らかになり,神経細胞移動機構の全体像が明らかとなることを期待する.
引用文献References
1) He, S., Li, Z., Ge, S., Yu, Y.C., & Shi, S.H. (2015) Neuron, 86, 1159–1166.
2) Gelman, D.M. & Marin, O. (2010) Eur. J. Neurosci., 31, 2136–2141.
3) Hevner, R.F., Daza, R.A., Englund, C., Kohtz, J., & Fink, A. (2004) Neuroscience, 124, 605–618.
4) Marin, O., Valiente, M., Ge, X., & Tsai, L.H. (2010) Cold Spring Harb. Perspect. Biol., 2, a001834.
5) Rakic, P. (1972) J. Comp. Neurol., 145, 61–83.
6) Nadarajah, B., Brunstrom, J.E., Grutzendler, J., Wong, R.O., & Pearlman, A.L. (2001) Nat. Neurosci., 4, 143–150.
7) Tabata, H. & Nakajima, K. (2003) J. Neurosci., 23, 9996–10001.
8) Sekine, K., Honda, T., Kawauchi, T., Kubo, K., & Nakajima, K. (2011) J. Neurosci., 31, 9426–9439.
9) Kato, M. (2015) Front. Neurosci., 9, 181.
10) Reiner, O., Carrozzo, R., Shen, Y., Wehnert, M., Faustinella, F., Dobyns, W.B., Caskey, C.T., & Ledbetter, D.H. (1993) Nature, 364, 717–721.
11) Hattori, M., Adachi, H., Tsujimoto, M., Arai, H., & Inoue, K. (1994) Nature, 370, 216–218.
12) Honda, A., Ono, J., Kurahashi, H., Mano, T., Imai, K., & Okada, S. (1998) Brain Dev., 20, 190–192.
13) Toyo-oka, K., Shionoya, A., Gambello, M.J., Cardoso, C., Leventer, R., Ward, H.L., Ayala, R., Tsai, L.H., Dobyns, W., Ledbetter, D., Hirotsune, S., & Wynshaw-Boris, A. (2003) Nat. Genet., 34, 274–285.
14) Faulkner, N.E., Dujardin, D.L., Tai, C.Y., Vaughan, K.T., O’Connell, C.B., Wang, Y., & Vallee, R.B. (2000) Nat. Cell Biol., 2, 784–791.
15) Hirotsune, S., Fleck, M.W., Gambello, M.J., Bix, G.J., Chen, A., Clark, G.D., Ledbetter, D.H., McBain, C.J., & Wynshaw-Boris, A. (1998) Nat. Genet., 19, 333–339.
16) Hong, S.E., Shugart, Y.Y., Huang, D.T., Shahwan, S.A., Grant, P.E., Hourihane, J.O., Martin, N.D., & Walsh, C.A. (2000) Nat. Genet., 26, 93–96.
17) Falconer, D.S. (1951) J. Genet., 50, 192–201.
18) D’Arcangelo, G. (2005) Int. Rev. Neurobiol., 71, 383–417.
19) D’Arcangelo, G., Miao, G.G., Chen, S.C., Soares, H.D., Morgan, J.I., & Curran, T. (1995) Nature, 374, 719–723.
20) D’Arcangelo, G., Nakajima, K., Miyata, T., Ogawa, M., Mikoshiba, K., & Curran, T. (1997) J. Neurosci., 17, 23–31.
21) Ogawa, M., Miyata, T., Nakajima, K., Yagyu, K., Seike, M., Ikenaka, K., Yamamoto, H., & Mikoshiba, K. (1995) Neuron, 14, 899–912.
22) Miyata, T., Nakajima, K., Aruga, J., Takahashi, S., Ikenaka, K., Mikoshiba, K., & Ogawa, M. (1996) J. Comp. Neurol., 372, 215–228.
23) Nakajima, K., Mikoshiba, K., Miyata, T., Kudo, C., & Ogawa, M. (1997) Proc. Natl. Acad. Sci. USA, 94, 8196–8201.
24) Pesold, C., Impagnatiello, F., Pisu, M.G., Uzunov, D.P., Costa, E., Guidotti, A., & Caruncho, H.J. (1998) Proc. Natl. Acad. Sci. USA, 95, 3221–3226.
25) Uchida, T., Baba, A., Perez-Martinez, F.J., Hibi, T., Miyata, T., Luque, J.M., Nakajima, K., & Hattori, M. (2009) J. Neurosci., 29, 10653–10662.
26) Perez-Martinez, F.J., Luque-Rio, A., Sakakibara, A., Hattori, M., Miyata, T., & Luque, J.M. (2012) Biol. Open, 1, 1258–1263.
27) Yasui, N., Nogi, T., Kitao, T., Nakano, Y., Hattori, M., & Takagi, J. (2007) Proc. Natl. Acad. Sci. USA, 104, 9988–9993.
28) D’Arcangelo, G., Homayouni, R., Keshvara, L., Rice, D.S., Sheldon, M., & Curran, T. (1999) Neuron, 24, 471–479.
29) Hiesberger, T., Trommsdorff, M., Howell, B.W., Goffinet, A., Mumby, M.C., Cooper, J.A., & Herz, J. (1999) Neuron, 24, 481–489.
30) Bock, H.H. & Herz, J. (2003) Curr. Biol., 13, 18–26.
31) Howell, B.W., Herrick, T.M., & Cooper, J.A. (1999) Genes Dev., 13, 643–648.
32) Trommsdorff, M., Gotthardt, M., Hiesberger, T., Shelton, J., Stockinger, W., Nimpf, J., Hammer, R.E., Richardson, J.A., & Herz, J. (1999) Cell, 97, 689–701.
33) Howell, B.W., Hawkes, R., Soriano, P., & Cooper, J.A. (1997) Nature, 389, 733–737.
34) Sheldon, M., Rice, D.S., D’Arcangelo, G., Yoneshima, H., Nakajima, K., Mikoshiba, K., Howell, B.W., Cooper, J.A., Goldowitz, D., & Curran, T. (1997) Nature, 389, 730–733.
35) Howell, B.W., Herrick, T.M., Hildebrand, J.D., Zhang, Y., & Cooper, J.A. (2000) Curr. Biol., 10, 877–885.
36) Kuo, G., Arnaud, L., Kronstad-O’Brien, P., & Cooper, J.A. (2005) J. Neurosci., 25, 8578–8586.
37) Bock, H.H., Jossin, Y., Liu, P., Forster, E., May, P., Goffinet, A.M., & Herz, J. (2003) J. Biol. Chem., 278, 38772–38779.
38) Beffert, U., Morfini, G., Bock, H.H., Reyna, H., Brady, S.T., & Herz, J. (2002) J. Biol. Chem., 277, 49958–49964.
39) Ballif, B.A., Arnaud, L., Arthur, W.T., Guris, D., Imamoto, A., & Cooper, J.A. (2004) Curr. Biol., 14, 606–610.
40) Chen, K., Ochalski, P.G., Tran, T.S., Sahir, N., Schubert, M., Pramatarova, A., & Howell, B.W. (2004) J. Cell Sci., 117, 4527–4536.
41) Huang, Y., Magdaleno, S., Hopkins, R., Slaughter, C., Curran, T., & Keshvara, L. (2004) Biochem. Biophys. Res. Commun., 318, 204–212.
42) Feng, L., Allen, N.S., Simo, S., & Cooper, J.A. (2007) Genes Dev., 21, 2717–2730.
43) Kubo, K., Mikoshiba, K., & Nakajima, K. (2002) Neurosci. Res., 43, 381–388.
44) Nakano, Y., Kohno, T., Hibi, T., Kohno, S., Baba, A., Mikoshiba, K., Nakajima, K., & Hattori, M. (2007) J. Biol. Chem., 282, 20544–20552.
45) Strasser, V., Fasching, D., Hauser, C., Mayer, H., Bock, H.H., Hiesberger, T., Herz, J., Weeber, E.J., Sweatt, J.D., Pramatarova, A., Howell, B., Schneider, W.J., & Nimpf, J. (2004) Mol. Cell. Biol., 24, 1378–1386.
46) Kohno, T., Nakano, Y., Kitoh, N., Yagi, H., Kato, K., Baba, A., & Hattori, M. (2009) J. Neurosci. Res., 87, 3043–3053.
47) Lambert de Rouvroit, C., de Bergeyck, V., Cortvrindt, C., Bar, I., Eeckhout, Y., & Goffinet, A.M. (1999) Exp. Neurol., 156, 214–217.
48) Jossin, Y., Ignatova, N., Hiesberger, T., Herz, J., Lambert de Rouvroit, C., & Goffinet, A.M. (2004) J. Neurosci., 24, 514–521.
49) Jossin, Y., Gui, L., & Goffinet, A.M. (2007) J. Neurosci., 27, 4243–4252.
50) Kohno, T., Honda, T., Kubo, K., Nakano, Y., Tsuchiya, A., Murakami, T., Banno, H., Nakajima, K., & Hattori, M. (2015) J. Neurosci., 35, 4776–4787.
51) Koie, M., Okumura, K., Hisanaga, A., Kamei, T., Sasaki, K., Deng, M., Baba, A., Kohno, T., & Hattori, M. (2014) J. Biol. Chem., 289, 12922–12930.
52) Sato, Y., Kobayashi, D., Kohno, T., Kidani, Y., Prox, J., Becker-Pauly, C., & Hattori, M. (2015) J. Biochem., in press.
53) Kohno, S., Kohno, T., Nakano, Y., Suzuki, K., Ishii, M., Tagami, H., Baba, A., & Hattori, M. (2009) Biochem. Biophys. Res. Commun., 380, 93–97.
54) Hisanaga, A., Morishita, S., Suzuki, K., Sasaki, K., Koie, M., Kohno, T., & Hattori, M. (2012) FEBS Lett., 586, 3349–3353.
55) Krstic, D., Rodriguez, M., & Knuesel, I. (2012) PLoS ONE, 7, e47793.
56) Trotter, J.H., Lussier, A.L., Psilos, K.E., Mahoney, H.L., Sponaugle, A.E., Hoe, H.S., Rebeck, G.W., & Weeber, E.J. (2014) Neuroscience, 274, 299–307.
57) Hibi, T., Mizutani, M., Baba, A., & Hattori, M. (2009) Neurosci. Res., 63, 251–258.
58) Kubo, K., Honda, T., Tomita, K., Sekine, K., Ishii, K., Uto, A., Kobayashi, K., Tabata, H., & Nakajima, K. (2010) J. Neurosci., 30, 10953–10966.
59) Sekine, K., Kawauchi, T., Kubo, K., Honda, T., Herz, J., Hattori, M., Kinashi, T., & Nakajima, K. (2012) Neuron, 76, 353–369.
60) Tabata, H. & Nakajima, K. (2002) J. Neurosci. Res., 69, 723–730.
61) Olson, E.C., Kim, S., & Walsh, C.A. (2006) J. Neurosci., 26, 1767–1775.
62) Franco, S.J., Martinez-Garay, I., Gil-Sanz, C., Harkins-Perry, S.R., & Muller, U. (2011) Neuron, 69, 482–497.
63) Jossin, Y. & Cooper, J.A. (2011) Nat. Neurosci., 14, 697–703.
64) Gil-Sanz, C., Franco, S.J., Martinez-Garay, I., Espinosa, A., Harkins-Perry, S., & Muller, U. (2013) Neuron, 79, 461–477.
65) Niu, S., Renfro, A., Quattrocchi, C.C., Sheldon, M., & D’Arcangelo, G. (2004) Neuron, 41, 71–84.
66) Jossin, Y. & Goffinet, A.M. (2007) Mol. Cell. Biol., 27, 7113–7124.
67) O’Dell, R.S., Cameron, D.A., Zipfel, W.R., & Olson, E.C. (2015) J. Neurosci., 35, 10659–10674.
68) Hirota, Y., Kubo, K., Katayama, K., Honda, T., Fujino, T., Yamamoto, T.T., & Nakajima, K. (2015) J. Comp. Neurol., 523, 463–478.
69) Hack, I., Hellwig, S., Junghans, D., Brunne, B., Bock, H.H., Zhao, S., & Frotscher, M. (2007) Development, 134, 3883–3891.
70) Gilmore, E.C. & Herrup, K. (2000) Curr. Biol., 10, R162–R166.
71) Dulabon, L., Olson, E.C., Taglienti, M.G., Eisenhuth, S., McGrath, B., Walsh, C.A., Kreidberg, J.A., & Anton, E.S. (2000) Neuron, 27, 33–44.
72) Sanada, K., Gupta, A., & Tsai, L.H. (2004) Neuron, 42, 197–211.
73) Belvindrah, R., Graus-Porta, D., Goebbels, S., Nave, K.A., & Muller, U. (2007) J. Neurosci., 27, 13854–13865.
74) Magdaleno, S., Keshvara, L., & Curran, T. (2002) Neuron, 33, 573–586.
75) Yoshida, M., Assimacopoulos, S., Jones, K.R., & Grove, E.A. (2006) Development, 133, 537–545.
76) Meyer, G., Cabrera Socorro, A., Perez Garcia, C.G., Martinez Millan, L., Walker, N., & Caput, D. (2004) J. Neurosci., 24, 9878–9887.
77) Lane-Donovan, C., Philips, G.T., Wasser, C.R., Durakoglugil, M.S., Masiulis, I., Upadhaya, A., Pohlkamp, T., Coskun, C., Kotti, T., Steller, L., Hammer, R.E., Frotscher, M., Bock, H.H., & Herz, J. (2015) Sci. Signal., 8, ra67.
著者紹介Author Profile
 河野 孝夫(こうの たかお)
河野 孝夫(こうの たかお)名古屋市立大学大学院薬学研究科助教.博士(薬学).
略歴1982年愛知県に生る.2005年名城大学薬学部卒業.07年名古屋市立大学大学院薬学研究科博士前期課程修了.10年同博士後期課程修了.08~10年日本学術振興会特別研究員DC2. 10年より現職.
研究テーマと抱負脳の形成と機能に必須なタンパク質リーリンの機能制御機構の解明.美しい脳の構造がいかにして形成,維持されるのかを明らかにし,精神神経疾患の発症機構の解明や,診断・治療法の開発につなげていきたい.
ウェブサイトhttp://www.phar.nagoya-cu.ac.jp/hp/bsk/indexj1.html
趣味音楽鑑賞,ギター演奏.
 服部 光治(はっとり みつはる)
服部 光治(はっとり みつはる)名古屋市立大学大学院薬学研究科教授.博士(薬学).
略歴1968年三重県に生る.91年東大薬学部卒.東大薬学部助手,ハーバード大学博士研究員,東大医科研助手を経て,2004年名古屋市立大学大学院薬学研究科独立助教授.09年より現職.