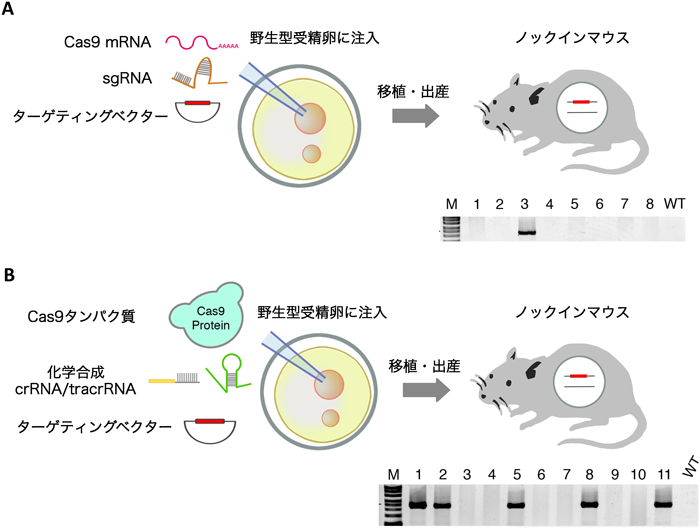
CRISPR/Casでマウスゲノムを自在に操るGenome editing in mouse with CRISPR/Cas system
東京医科歯科大学難治疾患研究所分子神経科学分野Laboratory of Molecular Neuroscience, Medical Research Institute, Tokyo Medical and Dental University ◇ 〒113–8510 東京都文京区湯島1–5–45 ◇ 1–5–45, Yushima, Bunkyo, Tokyo, 113–8510, Japan
発行日:2016年2月25日Published: February 25, 2016