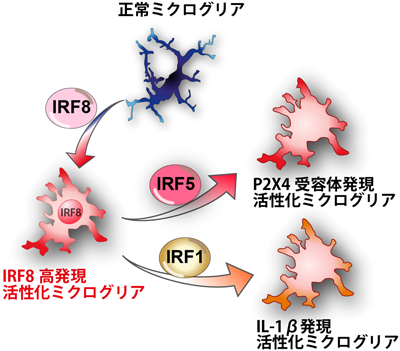
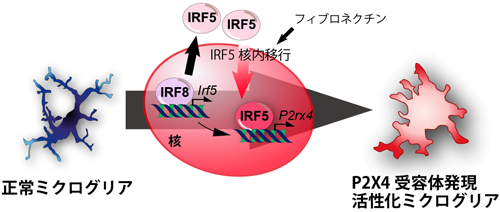
ミクログリアの活性化と形質を制御するIRF転写因子ファミリーRole of IRF transcription factor family in regulating reactive phenotype of microglia
1 フライブルク大学神経病理学研究所University of Freiburg, Institute of Neuropathology ◇ ドイツ フライブルク ブライザッカー通り64 神経センター ◇ Neurozentrum, Breisacherstr.64, D-79106 Freiburg, Germany
2 九州大学大学院薬学研究院ライフイノベーション分野Department of Life Innovation, Graduate School of Pharmaceutical Sciences, Kyushu University ◇ 福岡県福岡市東区馬出3–1–1 ◇ Maidashi 3–1–1, Higashi-ku, Fukuoka 812–8582, Japan
3 九州大学大学院薬学研究院薬理学分野Department of Molecular and System Pharmacology, Graduate School of Pharmaceutical Sciences, Kyushu University ◇ 福岡県福岡市東区馬出3–1–1 ◇ Maidashi 3–1–1, Higashi-ku, Fukuoka 812–8582, Japan
発行日:2016年2月25日Published: February 25, 2016