脳を他の臓器と比較した際目立つ特徴の一つは,それを構成する細胞の多様性と脳の部位によって異なる細胞の種類の多さである.特定の前駆細胞から生まれる神経細胞もその誕生時期に依存して異なるものが作られるが,脳の中にはそれだけでは説明できないほど多様な神経細胞が存在する部位もある.その典型例が小脳皮質であり,小脳皮質には6種類の異なる神経細胞(2種類の興奮性細胞と4種類の抑制性細胞)が存在する.これには異なる部位で生まれた神経細胞の移動の寄与が大きいが,そのような神経細胞の存在様式は神経回路形成に影響を及ぼし,ひいては神経回路の機能発現につながる.すなわち,脳における神経細胞移動はその機能発現に欠かすことのできない重要な現象である.
神経細胞は移動しても痕跡を残すわけではないため,固定標本の観察で伸長経路を追える軸索投射とは異なり,それを把握するのは容易ではない.無論,細胞を解離培養すれば試験管内での挙動の観察は可能であるが,脳の中での動きを捉えるには限界がある.古くからトリチウムチミジンなどを用いて特定の時期に分裂する細胞を捉え,その後の動きを観察するという方法も用いられてきたが,細胞を集団としてしか捉えることしかできず,また,分裂後長時間にわたる挙動を捉えることもできなかった.比較的最近になって用いられるようになったのが,DiIなどの脂溶性蛍光色素を用いる方法であったが,特定の時期に特定の部位に存在する細胞を非特異的に染めることしかできず,大まかな情報しか得られなかった上,細胞毒性の問題もあった.そのような理由で,神経回路形成の研究に比べて神経細胞移動の研究は大きく遅れていた.その後細胞標識や観察の技術の進展により,神経細胞移動の研究が大きく進展した.前者はGFP(green fluorescent protein)の出現や子宮内電気穿孔法,後者は共焦点顕微鏡や冷却CCDなどである.これらを組み合わせることにより,特定の時期に特定の部位から発生する神経細胞やその前駆体を生きたまま標識し,長期間にわたって観察することが可能となった.またさらに2光子顕微鏡を利用することで,生体中での神経細胞の移動を捉えることも可能となり,神経細胞移動の研究は大きく進展した.
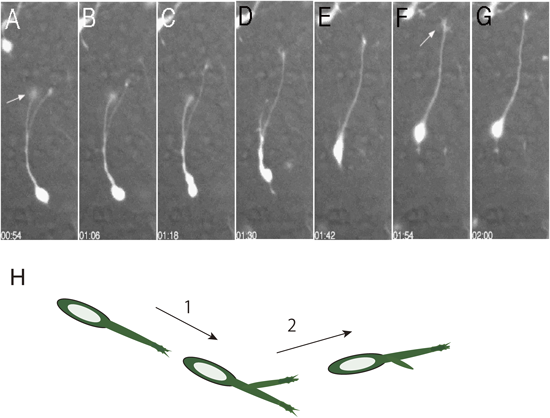
成熟脳における神経細胞は樹状突起と軸索と呼ばれる2種類の突起によって特徴づけられる.前者はシナプス入力を受け,後者は他の神経細胞にシナプスを形成する.一方,移動中の細胞は当然のことながらそのような突起を有さない.その代わりに,先導突起(leading process)と呼ばれる比較的太い突起を移動方向に伸ばし,後方にはトレーリングプロセス(trailing process)と呼ばれる短い突起を伸ばしながら移動する.突起の先端には軸索の成長円錐に似た小さな膨らみがある(図5参照).また,先導突起は状況によって枝分かれをする.移動速度は細胞の種類,発達の時期,環境によって異なるが,マウスの大脳皮質のGABA作動性介在ニューロンの場合10~30 µm/h1, 2)である.つまり,1日あたり250~700 µmも移動することになる.これは発達期の脳の大きさを考えるときわめて大きな距離である(移動速度が脳の大きさによるかどうかは知られていない).
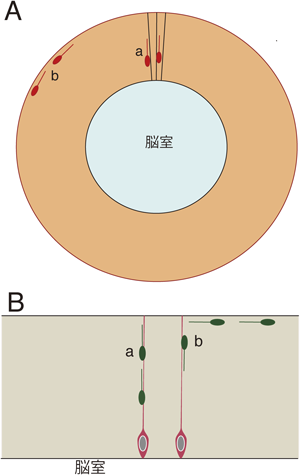
発達期の脳では二つの軸に沿った移動が起こる.その一つは神経管の中心と表面を結ぶ軸,すなわち法線方向への移動であり,これは神経管全体にわたって起こる(図1A).
1)神経管における法線方向への細胞移動
神経管の大部分の部位では神経細胞は脳室層で生まれた後,神経管の表面に向かって移動を開始する(図1).その方向は脳の表面に垂直であり,法線方向の移動(radial migration)と呼ばれる.大脳皮質でも,そこで生まれた神経細胞は法線方向の移動を開始する.しかし,他の部位にない特徴として,後から生まれた細胞が,先に生まれた細胞を追い越してより表面に位置するようになることがあげられる.その結果,II/III層からVI層にわたって分布する神経細胞は表面に近いほど遅生まれのものとなっている3).
2)神経管における接線方向への細胞移動
神経細胞には接線方向にも移動するものがある(tangential migration).そのような移動も脳の中で広く起こるが,大脳皮質,後脳,小脳の細胞の移動が比較的よく知られている.
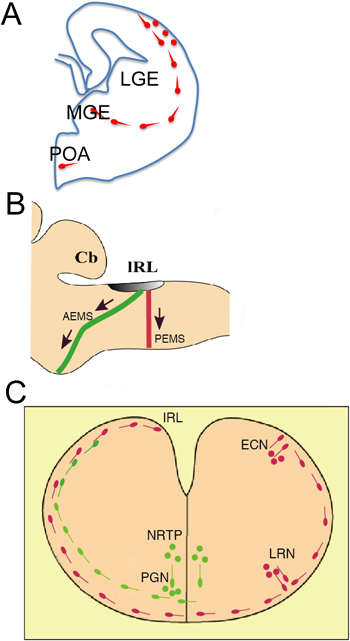
大脳皮質を構成する神経細胞は興奮性と抑制性に大別されるが,前者は大脳皮質の脳室付近で生まれて法線方向へ移動するが,後者は大脳皮質とは異なる部位で生まれ,長距離を移動して大脳皮質に到達する(図1Ab).すなわち,大脳皮質抑制性介在ニューロンの大部分は大脳皮質の腹側部の大脳基底核原基で発生し,接線方向に移動することにより,大脳皮質に到達する(図2A).実はこの事実が明らかになったのは比較的近年のことである.培養系にDiIを用いた染色を適用した実験,発達期脳の基底核原基に特異的に発現している転写因子Dlx1/Dlx2の発達に伴う発現パターンの変化,Dlx1/Dlx2のダブルノックアウトマウスの解析,脳の部分的切除実験などから,基底核原基が大脳皮質介在ニューロンの起源であることが示された4, 5).当初は外側基底核原基(lateral ganglionic eminence:LGE)がその起源と思われていたが,その後の研究で内側基底核原基(medial ganglionic eminence:MGE)が主たる起源であることが明らかになった6–8).また,MGE由来の細胞に少し遅れて尾側部の尾側基底核原基(caudal ganglionic eminence:CGE)からも介在ニューロンが発生することが明らかになった9–12).
後脳においても接線方向への著しい神経細胞移動が起こる(図2B, C).後脳は小脳前核細胞と呼ばれる細胞群が,神経核を形成する.これらの神経核は大脳や脊髄から入力を受け,小脳に興奮性入力を送っている.後脳においても大部分の神経細胞は法線方向に移動するが,一部は接線方向に移動する.後脳の最背側部に一過的に下菱脳唇(lRL)という薄く外から透けて見える構造が形成される.後脳における神経細胞の産生は比較的早い時期に起こるのに対して,この部位では後脳の他の部位での神経細胞産生が終わってからも神経細胞が作り続けられる.この部位で産生された神経細胞は脳表に沿って腹側に向かって接線方向に移動し,腹側部において神経核(小脳前核群)を形成する(図2B, C)13, 14).後脳にはこれ以外にも接線方向に移動する細胞がある.その一つが顔面神経核を構成する運動ニューロンで,この神経細胞は神経管の吻尾軸に沿って移動する15).
小脳前核群が産生される部位よりも吻側の最背側部に形成される上菱脳唇では,下菱脳唇と同様比較的発生の遅い時期まで神経細胞が産生されるが,この部位からは小脳顆粒細胞の前駆体や深部核の神経細胞が産生される.これらは髄膜直下を接線方向に移動していき,前者は外顆粒層を形成する16, 17).
このように,脳における神経細胞の移動はさまざまな系を用いて研究が進められてきたが,誕生後の神経細胞がいかにして最終目的地にたどり着き,成熟するかについて二つの系に焦点を当てて紹介する.
1)大脳皮質抑制性介在ニューロン
MGEから発生した介在ニューロンは法線方向に移動するが,その後接線方向に向きを変え,大脳皮質に向かって移動する(図2A).介在ニューロンの大半はMGEに由来するが約3割の細胞はより尾側に位置するCGEに由来する.また,約1割の介在ニューロンは尾腹側に位置する視索前野(prepotic area:POA)に由来するといわれている18).
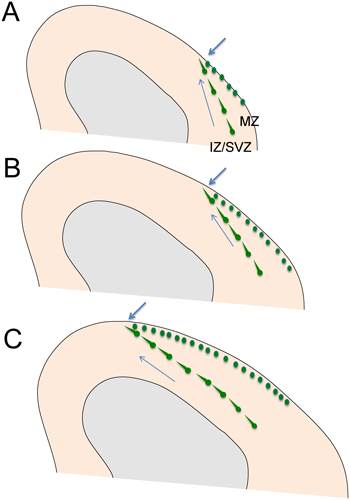
まず初めに,主たる起源であるMGEからの神経細胞移動について述べる.MGE由来の細胞はCGE由来の細胞に比べて早生まれであることから,GABAニューロン特異的にGFPを発現させたGAD67-GFPノックインマウス(以下GADマウスと呼ぶ)を用いて発生の早い時期に観察を行うことでその振る舞いを観察することができる1).また,MGEを構成する細胞の大部分は転写因子Nkx2.1を発現する前駆細胞に由来することから,Nkx2.1creマウスと適当なレポータマウスを組み合わせることでこれらを特異的に標識することができる19).また,子宮内電気穿孔法を用いてMGEに狙いを定めて蛍光タンパク質をコードする遺伝子を導入することで標識することも可能である20).これらの方法で標識したMGE由来の介在ニューロンは,マウスでは胎生12.5日ごろに大脳皮質の外側部から侵入する.その後,皮質では中間帯から脳室下帯にかけて皮質深部と辺縁帯に標識細胞が観察されるようになる(図3)1).このことは皮質の表面と深部の二つのルートを通って介在ニューロンが皮質に移動していくと考えることもできるが,この二つの部位の介在ニューロンは向きも動きもダイナミクスも大きく異なる.冠状断面で観察すると,中間帯/脳室下帯の介在ニューロンの多くは背内側に向いており,その方向に向かって移動していることがわかるのに対して,辺縁帯の介在ニューロンの方向性は明確ではなく,動きもほとんど確認できない.この一見奇妙な振る舞いに関して次のような可能性が考えられる.皮質板の形成は皮質内側では遅れることから,内側部では,中間帯/脳室下帯と辺縁帯が融合している.そのため中間帯/脳室下帯に分布する介在ニューロンと辺縁帯に分布する介在ニューロンは内側では合流している(図3矢印).すなわち,中間帯/脳室下帯を通ってきた介在ニューロンはその先端部では辺縁帯に到達すると考えれば説明可能である.そうだとすれば皮質板の発達に伴って中間帯/脳室下帯と辺縁帯融合部は内側に進んで行き,その結果として辺縁帯の介在ニューロンも内側に拡がると考えることができる.さらにこれに皮質板を横切る中間帯/脳室下帯から辺縁帯への移動も加わり,分布の中心が辺縁帯に移るものと考えられる.
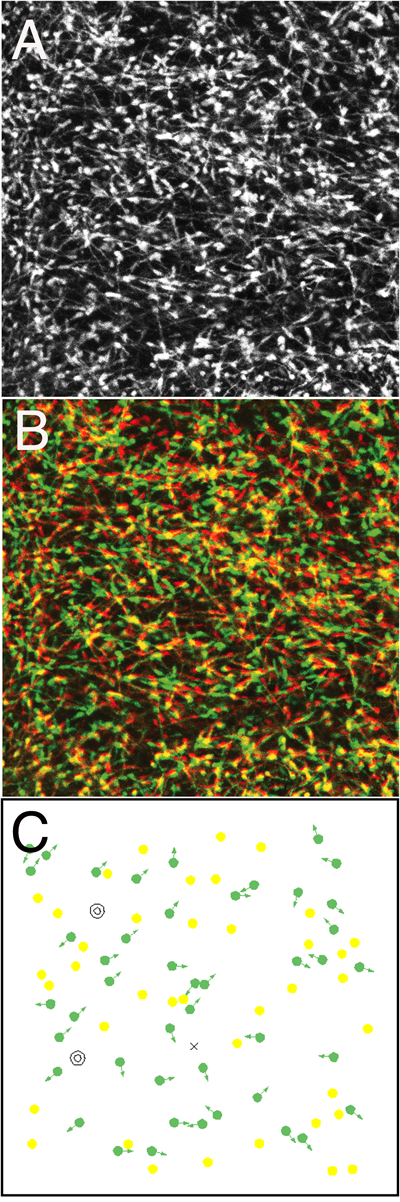
GADマウスの大脳皮質を切り取って表面から辺縁帯を観察すると,辺縁帯に到達したGFP陽性介在ニューロンは興味深い振る舞いを示す.すなわちこれらの介在ニューロンは不規則に多様な方向に向いて移動していることが確認できる(図4)1).GADマウスでは,すべての介在ニューロンが標識されてしまうため,個々の細胞の識別が困難であるが,子宮内電気穿孔法を用いると細胞を疎に標識することが可能となるため,個々の細胞の振る舞いを詳しく観察することができる.観察の結果,辺縁帯では介在ニューロンはランダムウォーク(random walk)様の動きをしていることが判明した21).ランダムウォークを続けると,細胞は皮質全体に拡がることが予想されるが,実際皮質の介在ニューロンは成熟脳では大脳皮質全体にほぼ均一に分布している.すなわち,MGEという特定の部位から発生した介在ニューロンは皮質辺縁帯においてランダムウォークをすることによって皮質全体への拡散が加速されると推測される.
MGE由来に比べて2日ほど遅れて発生するCGE由来の介在ニューロンも,やはり最初は中間帯・脳室下帯を通って移動し,その後辺縁帯に位置を変える.これらの細胞も辺縁帯でランダムウォークをするか否かは不明であるが,CGE由来の介在ニューロンも最終的に皮質に広く分布することを考えるとその可能性が高い.移動の初期には吻側に向かうとの報告もあるが22),その後いかにして皮質に拡がっていくかは明らかでない.
2)小脳前核細胞群
小脳前核細胞群のうち,橋核,橋被蓋網様核,外側網様核,楔状核に関してはトリチウムチミジンを用いた研究で古くから詳細に移動経路が調べられてきた13).これらの細胞はいずれも下菱脳唇で生まれた後,神経管の外周に沿って軟膜直下を接線方向に移動する(図2C).ただし,橋核(pontine nucleus:PN)と橋被蓋網様核(nucleus reticularis tegmenti pontis:NRTP)を形成する神経細胞は,吻側寄りのanterior extramural stream(AEMS)と呼ばれる経路を通り,腹側正中線付近へと移動する.外側網様核(lateral reticular nucleus:LRN)と外楔状核(external cuneatus nucleus:ECN)を形成する神経細胞は,尾側寄りのposterior extramural stream(PEMS)と呼ばれる別の経路を通り,やはり腹側正中線に向かって移動する(図2B).
トリチウムチミジンを用いると,細胞が両側性に標識されるため,正中線交差の有無に関する情報を得ることができないが,子宮内電気穿孔法を用いると片側の菱脳唇のみを標識することができるため,正中線を越える移動の有無を容易に確かめることができる.筆者らがこの方法を用いて調べた結果,PNとNRTPの細胞に関してはその一部(主としてNRTP)が正中線を越えて反対側に至り,残りは同側にとどまることが判明した23).一方LRNとECNを形成する細胞はすべてが正中線を越えて反対側に至り,ECNはより背側寄りに形成される.NRTP/PNの集合体の中で正中線付近で背側寄りの集合体は相対的に早生まれであり,両側に分布するが,腹側寄りの細胞は遅生まれであり,正中線を越えずに(起源となる菱脳唇と)同側にとどまっている23).
神経細胞が正しく目的地に到達し,移動を停止し,成熟するにはいくつかの重要な要素がある.
1)移動方向の転換
移動細胞の少なくとも一部は誘引性,反発性両方の軸索ガイダンス因子に対する受容体を発現しているが,培養系を用いることでこれらの因子に対する反応性を確認することができる24).これらのことは移動細胞も軸索の成長円錐と同様に化学物質の濃度勾配を検知して進行方向を変える能力を有していることを示している.
しかしながら,軸索の場合は先端に成長円錐と呼ばれる膨らみを用いて濃度勾配を検知するのに対して,移動細胞がどのようにして濃度勾配を検知するのかはよくわかっていない.先導突起の先端部の膨らみが軸索の成長円錐と同様な役割を果たす可能性も考えられるが,先導突起全体(運動性は先導突起全体にある)が濃度勾配検知装置として働く可能性も考えられる.
興味深いことに,移動中の細胞が移動方向を変えるときは先導突起の枝分かれが重要な役割を果たす.移動方向の転換は接線方向–法線方向の移動の切り替えのような直角の場合もあれば,より緩やかな角度の場合もある.まれにではあるが,180度の方向転換が観察されることもある.いずれの場合にも共通する点は,先導突起の枝分かれとそれに続く特定の分枝の選択によって方向を変えることができることである(図5).また,先導突起先端の膨らみは軸索成長円錐とは異なり,直進する傾向がある.このことは,先導突起の先端部だけではなく細胞全体が移動方向の転換に重要であることを示唆している.
2)移動経路の選択
これまでの研究から,移動経路を規定する要素は少なくとも二つあると考えられる.一つは物理的な基質であり,2番目は分子の局在である.前者の典型例はラディアルグリア(radial glia:RG)であり,大脳皮質興奮性ニューロンはRGの突起に沿って脳室層から脳表に向かって移動していくと考えられている(図1参照).外顆粒層に到達した小脳顆粒細胞の前駆細胞から生じた顆粒細胞は,RGに似た形態を示し,突起を髄膜まで伸ばすバーグマングリアと呼ばれる細胞の突起に沿って内顆粒層に向かって移動する25).またPN細胞も方向は逆であるが,RGの突起に沿って脳表から深部に移動する(図1B)23).後者の例としては,接線方向に移動する小脳前核細胞16),大脳皮質介在ニューロン1),カハールレチウス細胞26)や外顆粒層を移動する小脳顆粒細胞の前駆細胞16)があげられる.これらの細胞の移動経路には特定の構造は見あたらず,のちに詳述するように分子の局所的発現が経路を規定しているものと考えられる.
3)移動の終了
標的部位に到達した移動細胞は動きを止めて移動を終了しなければならない.しかしそのメカニズムに関しては多くは知られていない.基本的な問題として,内在的な機構によって制御されているのか,それとも外部からの要因によるのかという問題がある.
筆者の研究室では皮質介在ニューロンを用いてこの問題に取り組んだ.そのために同一の環境に生まれて間もない細胞と時間の経過した細胞を置き,その運動能を比較するというアプローチを採用した27).生体内に近い環境下での比較を行うため,大脳皮質の展開標本を用いた.そして同一個体を用いて胎生12.5日目にmCherryを,胎生15.5日目にgfpを電気穿孔法でMGEに導入することで,早生まれの細胞と遅生まれの細胞を標識した.胎生18.5目になると,いずれの細胞も辺縁帯に到達し,ランダムウォーク様の移動を始める.これを観察することにより,mCherryで赤く標識された細胞とGFPで緑に標識された介在ニューロンの運動能を同じ視野の中で比較することができる.その結果,赤く標識された細胞は緑に標識された細胞に比べて低い移動能を示した27).同一の環境下での比較であることを考えると,この移動能の違いは細胞自律的な要因によるものと考えられる.すなわち,細胞が生まれてから時間が経過するにつれ,皮質介在ニューロンは移動能が低下すると考えられる.
細胞自律的に運動能が制御されることは胎仔の神経細胞を成熟脳に移植した実験からも推測される.Wichterleらは胎仔脳から採取したMGEの組織を成熟マウスの脳に移植したところ,移植した細胞が脳の多様な部位に移動して拡がっていくことを見いだした8).つまり内在性の細胞の移動が起こらない成熟脳においても移動が起こったわけで,移動能は細胞自律的に制御されていることを示している.
むろん以上の結果は移動能を制御する環境因子の存在を否定するものではない.実際筆者らが胎生14.5日目の大脳皮質から採取した介在ニューロンが生後7日目の介在ニューロンから放出される因子にさらされる条件で培養を行ったところ,14.5日目の介在ニューロンの移動能が低下することを観察した27).
大脳皮質介在ニューロンが皮質の広い範囲にわたって分布するのに対して,小脳前核細胞は延髄の特定の部位に集合して神経核を形成する.また,大脳皮質介在ニューロンが生後1週間ごろになってようやく移動を終了するのに対して,小脳前核細胞は胎生期に移動を終了する.これらのことから小脳前核細胞はむしろ外部からの要因によって移動が停止する可能性が考えられる.
AEMSを移動するPNとNRTPを形成する細胞は神経管外周に沿って接線方向に移動したのち,一部は正中線の手前にとどまるが,一部は正中線を越えて移動し,反対側に集合体を形成する.一方ECN/LRNを形成する細胞はPEMSを移動し,すべて正中線を越えて反対側に到達してから,腹側寄り(LRN)と背側寄り(ECN)の2か所で塊を形成する(図2C).
これらのうち後者の振る舞いを制御する機構に関しては組織培養系を用いた研究により,以下のことが明らかにされている.腹側正中線の底板細胞には軸索誘引因子Netrin-1が発現しており,それにより,ECN/LRN細胞は正中線に誘引される28, 29).正中線を越えると,ECN/LRN細胞は誘引因子への反応性を失うと同時にその先にある翼板にある誘引活性に対する反応性を新たに獲得して,移動を続ける.特定の部位に集合するのは,接線方向へ移動するECN/LRN細胞に働きかける何らかの手がかりがその部位に存在することを示唆する29)が,もしそうであるなら,同側で核形成が起こっても不思議ではない.しかしそうならない理由としては,少なくとも二つの可能性が考えられる.一つ目は手がかりへの反応性を獲得するためには正中線を越える必要があるというもので,二番目は細胞が生まれてから一定の時間が経過する必要があるというものである.筆者らはこれらの可能性を検証するため,ECN/LRNに発現しているN-Cadherinの発現を抑制して移動速度を低下させた.その結果,元来なら反対側に形成されるはずのECN/LRNが同側に形成された30).このことからECN/LRNを形成する細胞は移動経路に存在する手がかりに反応することにより,移動を止めて核形成を行うものと推測される.すなわち通常は反対側に到達するころにようやく反応性を獲得するが,(N-Cadherinの発現が不十分なため)移動速度が低下すると,同側を通過する時点ですでに反応性を獲得するため,同側に核が形成されると考えると説明可能である.
AEMSを移動する細胞に関しても,後述のように腹側正中線に発現する因子が停止に関与している可能性が考えられる.
小脳前核細胞が最終位置に到達したとき,これらの細胞は単純に接線方向への移動を停止するわけではない.その振る舞いを詳しく観察すると,以下のようなことがわかる.最終位置付近ではこれらの細胞の多くは移動速度を低下させるとともに,先導突起に分岐が生じ,分岐した先導突起は法線方向に伸びる.そしてRGの突起に沿って脳表面から離れる方向に移動する(図1Bb).このように最終位置では移動方向の転換が起こる31).移動速度の低下が起こる結果,このような方向転換が可能になるのか,分枝形成・方向転換が速度の低下を招くのかは今のところ不明である.いずれにせよ,RGの突起は移動細胞の最終位置付近での移動に関しても重要な役割を果たしているようである.
神経細胞の移動のさまざまな局面を制御する分子機構に関しても断片的ではあるが知見が集積しつつある.
1)大脳皮質介在ニューロン
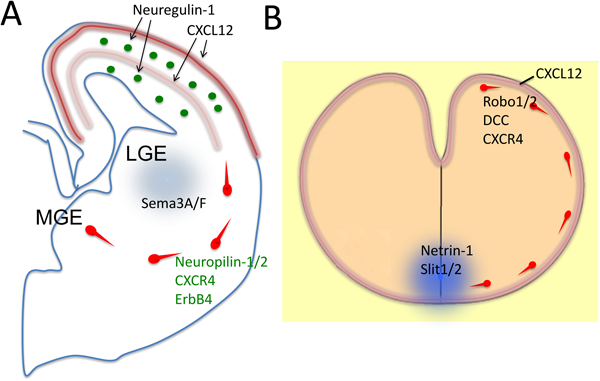
MGE由来の細胞は隣接するLGEには向かわず,その先にある皮質に向かう.その理由として反発性軸索ガイダンス因子Sema3(Semaphorin 3)の関与が考えられている.すなわち,LGEにはSema3A/Fが発現しているのに対して,介在ニューロンにはその受容体であるNeuropilin-1と2が発現している.これによりLGEから反発を受けるため,LGEには向かわないと考えられている(図6A)32).
また,皮質にはNeuregulinファミリーに属するNeuregulin-1の拡散性のアイソフォームであるNrg1-Igと,膜結合型のアイソフォームであるNrg1-CRDの両方が発現している.一方Neuregulin-1の受容体であるErbB4が移動中の皮質介在ニューロンに発現している.培養系ではNrg1-IgはMGE由来の細胞を誘引し,これらの細胞はNrg1-CRDを発現する細胞の基質を好む傾向がある.さらにNeuregulin/ErbB4シグナリングを阻害すると,皮質で観察される介在ニューロンの数が減少する.これらのことから大脳皮質介在ニューロンは皮質に発現するNeuregulinの活性によって皮質に誘引されるのではないかと考えられている(図6)33).
発達期の大脳皮質にはケモカインの一種であるCXCL12も発現しており,介在ニューロンはその受容体であるCXCR4も発現している.CXCL12は介在ニューロンの移動経路である中間帯・脳室下帯や辺縁帯に発現していることや,CXCL12が誘引活性を示す21, 34)ことから,CXCL12の発現が大脳皮質介在ニューロンの移動経路決定を規定している可能性が考えられている.実際CXCR4のノックアウトマウスでは中間帯・脳室下帯や辺縁帯における移動中の介在ニューロンの局在は著しく散漫なものになる21, 34, 35).これらのことから移動中の介在ニューロンの中間帯・脳室下帯や辺縁帯への局在にはCXCR4/CXCL12が関与していると考えられる(図6).
MGE由来の介在ニューロンは辺縁帯でランダムウォーク様の動きをするが,辺縁帯は介在ニューロンの最終分布部位ではない.これらの細胞はその後再び皮質板に移動して成熟する.移植実験などから,発生早期はCXCL12の影響でCXCR4を発現する介在ニューロンは皮質板に入れないが,後期になるとCXCL12/CXCR4シグナリングが弱まり,皮質板への進入が可能となると考えられている.その際,MGE由来の細胞は誕生時期依存的に早生まれの細胞ほど深部に分布する.その機構に関しては興奮性細胞の影響を受けているとの証拠がある.皮質の興奮性細胞も誕生時期依存的に早生まれの細胞ほど深部に分布し,リーリン,その受容体36–38),あるいはFezf2欠損マウス39)では興奮性細胞の層配置の異常が生ずるが,これらのマウスではそれに伴って介在ニューロンの層配置も乱れる.これらの事実は介在ニューロンの層配置が興奮性ニューロン由来の何らかのシグナルに依存している可能性を示している.
MGE由来の介在ニューロンが誕生時期依存的に皮質の特定の層に分布するのに対してCGE由来の介在ニューロンの層分布は誕生時期に依存しない.興味深いことにCGE由来の介在ニューロンの層分布決定には興奮性ニューロンの影響はあまり認められない39).
皮質介在ニューロンは長期間にわたって辺縁帯でランダムウォーク様の動きをするが,筆者らはその意義を調べるため,この時期にCXCL12/CXCR4シグナリングを抑制して,辺縁帯に介在ニューロンが滞在できないようにした.そしてマウスが成熟してから介在ニューロンの皮質内分布を調べたところ,少なくとも3種(Somatostatin-,Calretinin-,Neuropeptide Y)のマーカー分子を発現する介在ニューロンの層分布に変化が認められた40).このことは辺縁帯でランダムウォークをしている間に介在ニューロンが適切な層へ配置されるのに必要な能力の獲得をしていることを示唆している.
細胞の移動能の低下に関して,Bortoneらは生後発達期における介在ニューロンの運動能の低下がGABAの受容によって引き起こされるKイオントランスポーターKCC2のアップレギュレーションによることを示した41).このことは環境に存在するGABAの増加によって移動が抑制されうることを示している.
2)小脳前核細胞
小脳前核細胞は軟膜直下を通って接線方向へ移動する.その機構に関しては下記の結果が得られている.4種の小脳前核細胞はすべてケモカインCXCL12の受容体であるCXCR4を発現している.一方,髄膜にはCXCL12が発現している.培養下ではCXCL12発現細胞に向かって小脳前核細胞が移動していくことや,CXCL12やCXCR4のノックアウトマウスで髄膜から離れた位置を小脳前核細胞が移動していくことなどの事実から,小脳前核細胞は髄膜に発現するCXCL12によって誘引され続けることによって髄膜直下を移動していくものと考えられる(図6B)42).さらにCXCR4ノックアウトマウスにおいて,子宮内電気穿孔法を用いることで小脳前核細胞特異的にCXCR4を発現させると,ノックアウトマウスで認められる表現型が観察されなくなることから42),CXCR4は移動中の小脳前核細胞において細胞自律的に作用していることがわかる.
一方腹側正中線の底板には拡散性軸索誘引因子netrin-1が発現しているが,小脳前核細胞はNetrin-1の受容体の一つであるDCCを発現しており,培養下では底板の組織片やnetrin-1発現細胞塊に向かって移動していく.また,netrin-1やdccのノックアウトマウスでは,正中線に向かう移動が阻害される.これらのことから小脳前核細胞の正中線へ向かう移動にはNetrin-1の誘引活性が関与していると考えられる.LRNやECNを形成する細胞が正中線を越えてさらに移動していくメカニズムに関しては,これらの細胞がいったん底板に触れるとNetrin-1の誘引活性に対する反応性を失うのと同時に翼板の誘引活性(分子機構は不明である)に対する反応性を新たに獲得することによるとの知見が得られている29).さらに,後述のように腹側正中線には反発性ガイダンス因子Slitも発現しており,Robo/Slitシグナリングも小脳前核細胞の正中線交差に関係している.
AEMSを移動する細胞のうち,PN細胞は正中線を越えずに同側にとどまる.下オリーブ核細胞は小脳前核群よりもやや腹側で誕生するが,PN細胞と同様に接線方向に移動したのちに同側にとどまる.興味深いことに,PN細胞も下オリーブ核細胞も先導突起は正中線を越えて反対側に侵入する.このことは移動細胞の細胞体と先導突起の振る振る舞いが別々に制御されていることを示唆する.AEMSを移動する細胞の多くは同側にとどまるが,Slitの受容体であるRoboファミリータンパク質の発現パターンや,robo3のノックアウトマウスの解析から,この理由について次のように考えられている.これらの細胞はRobo3を発現しているが,移動に伴い先導突起が底板に触れることにより,Robo3の発現低下が起こり,その結果としてRobo2の発現が上昇し(Robo3は直接にはSlitの反発活性を受けないが,Robo1, Robo2の発現を抑制することでSlitへの反応性を制御する),正中線に発現するSlit1/2によって反発を受けることになり,正中線を越えることができず,同側にとどまるというものである43).AEMSを移動する細胞のうち正中線を交差する細胞やPEMSを移動する細胞に関しては,先導突起が短いためにRobo2の発現上昇が十分に起こる前に正中線を交差するものと考えられている.
過去10~15年の間に神経細胞移動の動態や分子機構の理解は大きく進展した.しかしながら,基本的な問題が未解明なまま残されている.たとえば移動の方向を決める際,拡散性因子の濃度勾配を検知するのは間違いなさそうであるが,それはどのようにして行うのか.検知部位は先導突起の先端なのか,先導突起全体なのか.また先導突起と細胞体の振る舞いは必ずしも一致しないことなどから,両者の動きはある程度独立に制御されている可能性が考えられるが,それはそのような機構によるものなのか.さらに移動の開始や終了はどのようにして制御されているのか.皮質介在ニューロンに関してはGABAが移動能の低下に関与している証拠が示されているが41),興奮性細胞の移動の停止はいかなる機構によるのか.また移動を終了した神経細胞は軸索を伸ばし始めるが44),それは何がトリガーとなり,どのようにして制御されているのであろうか.神経細胞移動の研究はまだ始まったところというべきかもしれない.
引用文献References
1) Tanaka, D., Nakaya, Y., Yanagawa, Y., Obata, K., & Murakami, F. (2003) Development, 130, 5803–5813.
2) Yanagida, M., Miyoshi, R., Toyokuni, R., Zhu, Y., & Murakami, F. (2012) Proc. Natl. Acad. Sci. USA, 109, 16737–16742.
3) Angevine, J.B. Jr. & Sidman, R.L. (1961) Nature, 192, 766–768.
4) Anderson, S.A., Eisenstat, D.D., Shi, L., & Rubenstein, J.L.R. (1997) Science, 278, 474–476.
5) Tamamaki, N., Fujimori, K.E., & Takauji, R. (1997) J. Neurosci., 17, 8313–8323.
6) Sussel, L., Marín, O., Kimura, S., & Rubenstein, J.L. (1999) Development, 126, 3359–3370.
7) Lavdas, A.A., Grigoriou, M., Pachnis, V., & Parnavelas, J.G. (1999) J. Neurosci., 19, 7881–7888.
8) Wichterle, H., Garcia-Verdugo, J.M., Herrera, D.G., & Alvarez-Buylla, A. (1999) Nat. Neurosci., 2, 461–466.
9) Wichterle, H., Turnbull, D.H., Nery, S., Fishell, G., & Alvarez-Buylla, A. (2001) Development, 128, 3759–3771.
10) Nery, S., Fishell, G., & Corbin, J.G. (2002) Nat. Neurosci., 5, 1279–1287.
11) Miyoshi, G., Hjerling-Leffler, J., Karayannis, T., Sousa, V.H., Butt, S.J., Battiste, J., Johnson, J.E., Machold, R.P., & Fishell, G. (2010) J. Neurosci., 30, 1582–1594.
12) Rubin, A.N., Alfonsi, F., Humphreys, M.P., Choi, C.K., Rocha, S.F., & Kessaris, N. (2010) J. Neurosci., 30, 12050–12062.
13) Altman, J. & Bayer, S.A. (1987) J. Comp. Neurol., 257, 529–552.
14) Altman, J. & Bayer, S.A. (1987) J. Comp. Neurol., 257, 513–528.
15) Chandrasekhar, A. (2004) Dev. Dyn., 229, 143–161.
16) Machold, R. & Fishell, G. (2005) Neuron, 48, 17–24.
17) Wang, V.Y., Rose, M.F., & Zoghbi, H.Y. (2005) Neuron, 48, 31–43.
18) Gelman, D.M. & Marín, O. (2010) Eur. J. Neurosci., 31, 2136–2141.
19) Xu, Q., Tam, M., & Anderson, S.A. (2008) J. Comp. Neurol., 506, 16–29.
20) Tanaka, D.H., Maekawa, K., Yanagawa, Y., Obata, K., & Murakami, F. (2006) Development, 133, 2167–2176.
21) Tanaka, D.H., Yanagida, M., Zhu, Y., Mikami, S., Nagasawa, T., Miyazaki, J., Yanagawa, Y., Obata, K., & Murakami, F. (2009) J. Neurosci., 29, 1300–1311.
22) Yozu, M., Tabata, H., & Nakajima, K. (2005) J. Neurosci., 25, 7268–7277.
23) Kawauchi, D., Taniguchi, H., Watanabe, H., Saito, T., & Murakami, F. (2006) Development, 133, 1113–1123.
24) Ward, M.E., Jiang, H., & Rao, Y. (2005) Mol. Cell. Neurosci., 30, 378–387.
25) Rakic, P. (1971) J. Comp. Neurol., 141, 283–312.
26) Takiguchi-Hayashi, K., Sekiguchi, M., Ashigaki, S., Takamatsu, M., Hasegawa, H., Suzuki-Migishima, R., Yokoyama, M., Nakanishi, S., & Tanabe, Y. (2004) J. Neurosci., 24, 2286–2295.
27) Inamura, N., Kimura, T., Tada, S., Kurahashi, T., Yanagida, M., Yanagawa, Y., Ikenaka, K., & Murakami, F. (2012) J. Neurosci., 32, 6032–6042.
28) Alcantara, S., Ruiz, M., De Castro, F., Soriano, E., & Sotelo, C. (2000) Development, 127, 1359–1372.
29) Taniguchi, H., Tamada, A., Kennedy, T.E., & Murakami, F. (2002) Dev. Biol., 249, 321–332.
30) Taniguchi, H., Kawauchi, D., Nishida, K., & Murakami, F. (2006) Development, 133, 1923–1931.
31) Watanabe, H. & Murakami, F. (2009) Neurosci. Res., 64, 20–29.
32) Marín, O., Yaron, A., Bagri, A., Tessier-Lavigne, M., & Rubenstein, J.L. (2001) Science, 293, 872–875.
33) Flames, N., Long, J.E., Garratt, A.N., Fischer, T.M., Gassmann, M., Birchmeier, C., Lai, C., Rubenstein, J.L., & Marín, O. (2004) Neuron, 44, 251–261.
34) Stumm, R.K., Zhou, C., Ara, T., Lazarini, F., Dubois-Dalcq, M., Nagasawa, T., Hollt, V., & Schulz, S. (2003) J. Neurosci., 23, 5123–5130.
35) Tiveron, M.C., Rossel, M., Moepps, B., Zhang, Y.L., Seidenfaden, R., Favor, J., Konig, N., & Cremer, H. (2006) J. Neurosci., 26, 13273–13278.
36) Hammond, V., So, E., Gunnersen, J., Valcanis, H., Kalloniatis, M., & Tan, S.S. (2006) J. Neurosci., 26, 1646–1655.
37) Hevner, R.F., Daza, R.A.M., Englund, C., Kohtz, J., & Fink, A. (2004) Neuroscience, 124, 605–618.
38) Pla, R., Borrell, V., Flames, N., & Marín, O. (2006) J. Neurosci., 26, 6924–6934.
39) Lodato, S., Rouaux, C., Quast, K.B., Jantrachotechatchawan, C., Studer, M., Hensch, T.K., & Arlotta, P. (2011) Neuron, 69, 763–779.
40) Tanaka, D.H., Mikami, S., Nagasawa, T., Miyazaki, J., Nakajima, K., & Murakami, F. (2010) Cereb. Cortex, 20, 2810–2817.
41) Bortone, D. & Polleux, F. (2009) Neuron, 62, 53–71.
42) Zhu, Y., Matsumoto, T., Mikami, S., Nagasawa, T., & Murakami, F. (2009) Development, 136, 1919–1928.
43) Marillat, V., Sabatier, C., Failli, V., Matsunaga, E., Sotelo, C., Tessier-Lavigne, M., & Chedotal, A. (2004) Neuron, 43, 69–79.
44) Yamasaki, E., Tanaka, D.H., Yanagawa, Y., & Murakami, F. (2010) J. Neurosci., 30, 1521.
著者紹介Author Profile
 村上 富士夫(むらかみ ふじお)
村上 富士夫(むらかみ ふじお)大阪大学大学院生命機能研究科名誉教授.工学博士.
略歴1948年愛知県生まれ.71年大阪大学基礎工学部卒業,同助手,助教授を経て88年基礎工学部教授.2002年大阪大学大学院生命機能研究科教授.2013年大阪大学名誉教授.09年よりさきがけ総括.
研究テーマと抱負最初に興味を抱いたのは神経回路形成の可塑性.その後,神経発生,特に神経回路形成,神経細胞移動の研究へと研究を展開した.分子機構の研究の前に現象を丁寧に観察することが重要と考えている.
趣味音楽鑑賞.