ヘム代謝系関連酵素の構造生物学的研究Structural study of enzymes involved in heme metabolism
久留米大学医学部Kurume University School of Medicine ◇ 〒830–0011 福岡県久留米市旭町67 ◇ 67 Asahi-machi, Kurume Fukuoka 830–0011, Japan
発行日:2016年4月25日Published: April 25, 2016
ヘムは生命活動に必須な補欠分子族であるが,タンパク質から遊離したヘムは活性酸素を生じる有害な分子となるので,速やかに除去する必要がある.この系はヘム代謝系として知られており,ヘムオキシゲナーゼ(HO)によって,ヘムはビリベルジン,一酸化炭素,鉄へと分解される.この系は生命に必須な鉄の供給源として重要であり,シグナル伝達物質としての機能が示唆されている一酸化炭素の発生源でもある.ビリベルジンは,哺乳類ではビリベルジン還元酵素によって,抗酸化物質として機能するビリルビンへと転換され,植物などの光合成生物では,フェレドキシン依存性ビリン還元酵素(FDBR)によって,さまざまな光合成色素や光受容色素へと転換される.筆者らはHOとHO反応に必要な還元力を供給するシトクロムP450還元酵素,およびFDBRの一種であるPcyAの立体構造と機能に関する研究を行った.その成果について紹介する.
© 2016 公益社団法人日本生化学会© 2016 The Japanese Biochemical Society
ヘムは中心金属として鉄を持つポルフィリン錯体で,酸化還元によって鉄が二価と三価を行き来する性質や二価鉄に酸素分子が結合する性質を利用することによって,電子伝達,酸素の運搬,酵素の活性中心として用いられる補欠分子族である.特に酸素呼吸を行うために必須な呼吸鎖を構成するシトクロム類はヘムタンパク質であり,ヘムは酸素呼吸を行うすべての生物の生命維持に欠かすことのできない分子である.しかし,その有用性はタンパク質に結合している場合に限られ,ヘムタンパク質の新陳代謝などによって生じた遊離のヘムは,酸素分子と化学反応を起こすことで活性酸素種を生じる有害な分子(プロオキシダント)である.したがって,遊離のヘムは速やかに分解される必要がある.この役割を担っているのが,ヘム代謝系である.
哺乳類において,ヘムを最も多量に含んでいるのは赤血球である.赤血球は120日周期の新陳代謝サイクルを持ち,老化した赤血球から生じる多量のヘモグロビンを速やかに分解する必要がある.老化赤血球は細網内皮系によって分解を受ける.このときに生じる遊離ヘムはヘムオキシゲナーゼ(heme oxygenase:HO)によって,ビリベルジン,二価鉄,一酸化炭素へと分解される(図1)1–3).二価鉄はトランスフェリンによる輸送やフェリチンによる貯蔵を経たのちに,最終的には骨髄でのヘムの合成などに再利用される.この経路によって供給される鉄は,1日に必要とされる鉄の90%以上を占めており,鉄の恒常性維持に非常に重要である.ビリベルジンはニコチンアミドアデニンジヌクレオチド/ニコチンアミドアデニンジヌクレオチドリン酸(NADH/NADPH)依存的なビリベルジン還元酵素によって,ビリルビンへと還元される(図1)4).ビリルビンは不溶性であるので,グルクロン酸抱合を受けて,可溶化され,胆汁中に排泄される.ビリルビン代謝に関する遺伝病(ジルベール症候群など)はいくつか報告されており,過剰なビリルビンの蓄積は黄疸となって現れる.ただし,生理的な濃度でのビリルビンはα-トコフェロールと同様に脂溶性の抗酸化剤として機能すると示唆されている5).この代謝経路はプロオキシダントであるヘムを分解し,抗酸化剤であるビリルビンを生成することや,HOの誘導型アイソザイムであるHO-1が,酸化ストレスや親電子性物質によってKeap1-Nrf2システムを介した発現誘導6)を受けることから,酸化ストレスに対する防御機構としても機能していると考えられている.一酸化炭素は一酸化窒素と同様にシグナル伝達分子として機能することが示唆されている.一酸化炭素の関与するシグナル伝達系としては,NF-κBを介した炎症や細胞増殖や分化7),シスタチオニンβ-シンターゼを介した血管拡張や収縮8, 9),NPAS2を介した体内時計10)などが示唆されている.
一方,植物や光合成生物においては,ヘム代謝系は光合成色素や光受容色素の合成に関与している.HO反応によって生じたビリベルジンはフェレドキシン依存性ビリン還元酵素(ferredoxin dependent bilin reductase:FDBR)によって還元を受け,フィコビリンやフィトクロモビリンを生じる(図1)11, 12).フィコビリンは紅藻やシアノバクテリアなどに存在する光合成色素で,これらの生物の光捕集器官であるフィコビリソーム中に含まれる.この色素はクロロフィルの吸収できない波長領域の光を吸収できるので,これらの生物では幅広い波長領域の光を使った光合成が可能となっている.フィトクロモビリンは植物に含まれる光受容色素で,赤色/遠赤色光受容体であるフィトクロムに含まれる発色団である.フィトクロモビリンがcis/trans光異性化反応を起こすことで,光受容に伴うフィトクロムの構造変化が誘起される.近年,シアノバクテリアや細菌でもフィコビリンやビリベルジンを発色団として使用するさまざまなフィトクロムホモログが発見されており,注目を集めている13, 14).
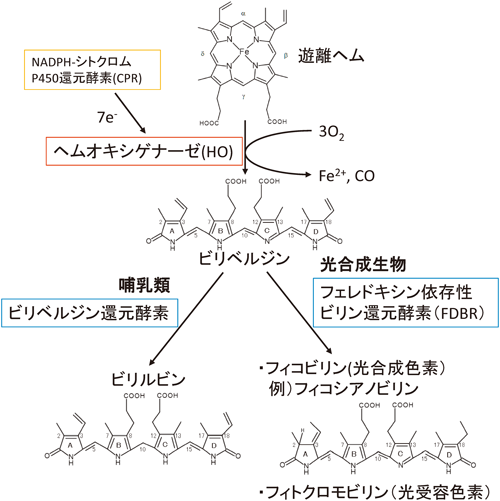
老化した赤血球の新陳代謝などによって生じた遊離ヘムはHOによって,ビリベルジン,鉄,一酸化炭素へと分解される.ビリベルジンは哺乳類ではビリベルジン還元酵素によって,ビリルビンへと代謝される.光合成生物ではFDBRによって,フィコビリンやフィトクロモビリンへと代謝される.
筆者らは,HOやFDBRの一種であるPcyAについて生化学・構造生物学的研究を進めてきた.本稿ではこれらの研究を中心に,周辺の研究についても概説する.近年,HOとはアミノ酸配列上,まったく相同性のないヘム分解酵素(IsdG, IsdI, MhuDなど)が細菌で発見され,立体構造も明らかにされているが,これらについては本稿では紹介しない.
哺乳類のHOはC末端側で小胞体膜に結合した膜タンパク質であり,小胞体膜にN末端側で結合したNADPH-シトクロムP450還元酵素(cytochrome P450 reductase:CPR)からの還元力を利用して,ヘムを分解する2, 3).この反応ではヘムはHOに基質として結合したのちに,還元力を受け取って,ヘム鉄に結合した酸素分子をヒドロペルオキシド(OOH−)へと活性化する.ヒドロペルオキシドはヘムポルフィリン環のα-メソ位を部位特異的に攻撃し,α-ヒドロキシヘムを生じる.すなわち,この過程において,ヘムは基質であると同時に補酵素としても機能している.HO反応の特徴は部位特異的ということであり,非酵素的反応では,ポルフィリンのすべてのメソ位のいずれかが切断される.α-ヒドロキシヘムはさらに酸化され,ベルドヘムへと変換される.この過程で,α-メソ位の炭素原子は一酸化炭素として放出される.その後,ポルフィリン環が切断され,ビリベルジン–鉄錯体となり,三価の鉄が二価に還元され,鉄の溶解度が上昇すると鉄が遊離し,ビリベルジンもHOから遊離する(図2a).
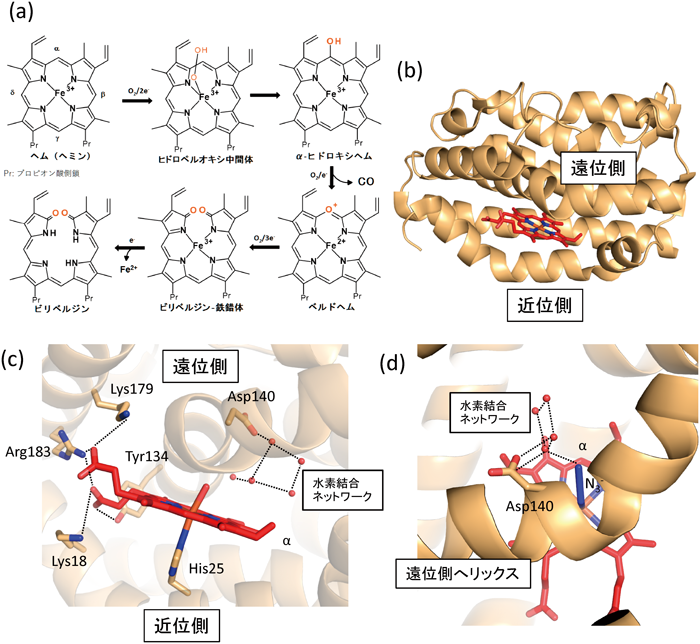
(a)HOの反応スキーム.HOはヘムのα-メソ位を部位特異的に開裂させ,ビリベルジンを生じる.(b)酸化型ヘム–HO-1複合体の全体構造.(c)酸化型ヘム–HO-1複合体のヘム周辺の立体構造.点線は水素結合または塩橋を示す.赤い球は水分子の酸素原子を示す.(d)アジド結合型ヘム–HO-1複合体の立体構造.
この反応機構の詳細について迫ろうと,カリフォルニア大学のThomas PoulosとPaul R. Ortiz de Montellanoらのグループ,東北大学の齋藤正男や山形大学の吉田匡らのグループ,筆者らの主に三つの研究グループが,由来の異なるHOを使って,構造生物学的研究を進めてきた.筆者らはラット由来HO-1を用いて,HO単体15),酸化型ヘム複合体16),還元型ヘム複合体17),酸素結合型ヘム複合体18),ベルドヘム複合体19),ビリベルジン–鉄錯体複合体20)のそれぞれについてX線結晶構造を決定してきた.以下では,これらの立体構造をもとに反応機構の詳細について説明する.
HO-1の立体構造として最初に決定されたものは,Poulosらのグループによるヒト由来HO-1の酸化型ヘム複合体である21).この立体構造は膜結合部分を切除したものであり,筆者らもその立体構造を使って,膜結合部分を切除したラット由来HO-1のヘム複合体の立体構造を決定した(図2b)16).現在までさまざまな反応状態の哺乳類HOの立体構造が明らかとなっているが,すべて膜結合部分を除いたものである(哺乳類以外のHOは膜タンパク質ではない).ラット由来HO-1とヒト由来HO-1の相同性は80%以上で,ほとんど立体構造に違いはなかったが,ヘム結合部位で違いがみられた.ヘムは近位側へリックス(ヘム鉄に配位結合しているヒスチジン残基を含む)と遠位側ヘリックスにはさまれて結合しているが,その遠位側ヘリックスの構造に違いがみられた.この違いは,後述するヘムがビリベルジン–鉄錯体へと分解されていく過程でみられた変化と同様で,遠位側へリックスの構造柔軟性を示していると考えている.さらに酸化型ヘム複合体ではヘム鉄の遠位側に水または水酸化物イオンが配位し,還元型ヘム複合体では遠位側の配位子がない(図2c).これらの違いにも関わらず,ラット由来HO-1の立体構造を比較する限りにおいては,遠位側へリックスや近位側へリックスの構造変化はみられなかった.
ヘムの結合方向は反応の部位特異性を規定する上で,重要である.ヘムには疎水的なポルフィリン環部位とγ-メソ位の方向に負の電荷を持つプロピオン酸側鎖がある.HOのヘム結合部位を観察すると,プロピオン酸側鎖はリシンやアルギニンとの塩橋やチロシンとの水素結合によって,安定化されており,ポルフィリン環側には疎水的な残基が多い(図2c).これらの特徴によって,ヘムの結合方向が規定されていると考えられた.実際に,これらのリシンやアルギニンの変異体では反応の部位特異性が変化することが確かめられた22).また,緑膿菌由来HOは哺乳類のHOとは反応の部位特異性が異なるが,プロピオン酸側鎖と相互作用していたリシンやアルギニンは保存されていない23).反応の部位特異性を規定するもう一つの構造的因子は,ヘム鉄に結合した酸素分子の結合方向である.HOの一段階目の反応では,酸素分子が活性化されたヒドロペルオキシドが求電子的にヘムのα-メソ炭素を攻撃する24).したがって,酸素分子はα-メソ炭素の方向を向いていると予想される.この点を明らかにするために,筆者らはヒドロペルオキシドと同様に酸化型ヘムに結合するアジド(N3−)をアナログとして,その結合方向をX線結晶構造解析で明らかにした25).その結果,アジドは予想どおりα-メソ炭素の方向を向いて結合していた(図2d).その原因は,①遠位側へリックスがヘムに非常に近く,他の方向を向くためには立体的障害が生じる.②αメソ炭素方向には複数の水分子から形成される水素結合ネットワークがあり,それらの水分子と水素結合することによって,α-メソ炭素の方向を向いたほうが安定化されるの二点によると考えられた.
その後,齋藤らのグループによって,酸素分子が還元型ヘムに結合した状態がジフテリア菌由来HOにおいて決定され,筆者らも同様の構造をラット由来HO-1を使って決定した18, 26).いずれも酸素分子は,α-メソ炭素方向を向いて結合するという結果であった(図3a).
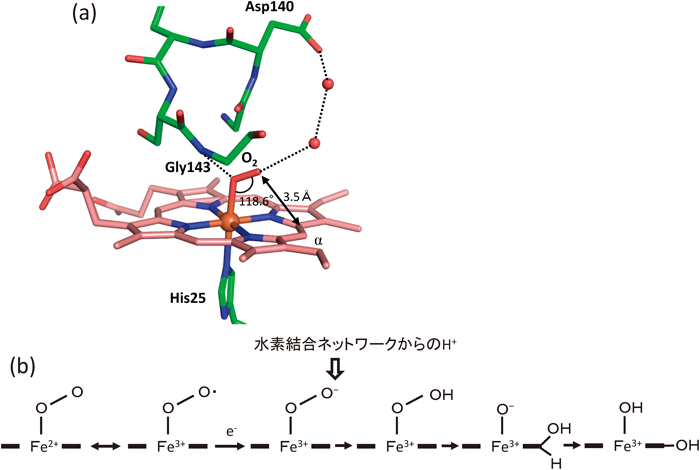
(a)ヘム周辺の立体構造.点線は水素結合を示す.ヘム複合体において水素結合ネットワークを形成していた五つの水分子は保存されているが,この図ではそのうちの2分子のみを示している.(b)酸素結合型からヒドロペルオキシ中間体を経由して,α-ヒドロキシヘムが生じるまでの反応機構.
上記の水素結合ネットワークは,酸素分子を活性化してヒドロペルオキシドへと転換する機構にも関与する.この水素結合ネットワークの末端にはAsp140があり,Asp140の変異体では活性が失われ27),水素結合ネットワークが不安定化することがわかっている28).酸素分子をヒドロペルオキシドにするためには,水素イオンを供給する必要があるが,その供給路としてこの水素結合ネットワークが機能しているのではないかと考えられている(図3b).現在では,これらの立体構造をもとにして,エルサレム・ヘブライ大学のSason Shaikや九州大学の吉澤一成らによって量子化学計算を使った研究が展開されており,上記の水素結合ネットワークの関与する反応機構の詳細が議論されている29, 30).
α-ヒドロキシヘムからベルドヘムへと至る過程では一酸化炭素が放出される.この反応過程における酸素と還元当量の必要性については,いまだに議論の分かれるところである31–34).α-ヒドロキシヘムもベルドヘムも酸素に対して不安定な分子であり,実験が非常にやりにくいためである.ただ筆者らは,この過程に関してはHOの関与は少なく,化学反応に近いと推定している35).
ベルドヘム複合体のX線結晶構造は,ヒト由来HO-1を用いてPoulosらのグループによって最初に明らかにされた36).ただ,この結晶構造中では分光学的に存在が示唆されていた第6配位子37, 38)やヘム複合体でみられていた水素結合ネットワークがみられないなどいろいろと問題点が指摘されていた.その後,筆者らはラット由来HO-1を用いて,齋藤らはジフテリア菌由来HOを用いて,ほぼ同時期にベルドヘム複合体の立体構造を報告した19, 39).筆者らは,ヘムとアスコルビン酸を反応させ,4種のアイソマーをHPLCで分画する方法でベルドヘムを調製し40),嫌気的にベルドヘム複合体を調製,結晶化した.この方法で得られた結晶構造には第6配位子や水素結合ネットワークがみられ,ほぼヘム複合体と同様の立体構造であった.
次の段階でベルドヘムのポルフィリン環は切断され,ビリベルジン–鉄錯体となる.筆者らはビリベルジン–鉄錯体複合体結晶を,ヘム複合体結晶にアスコルビン酸を加えて,結晶中で酵素反応を行わせることにより調製した(図4a)20).ベルドヘム→ビリベルジン–鉄錯体の過程では,おそらくヘム→α-ヒドロキシヘムと同様にベルドヘム鉄に酸素が結合し,そこに電子が供給されることによって,酸素分子がヒドロペルオキシドへと活性化され,反応が進行すると考えられる41).ベルドヘム複合体でも水素結合ネットワークは維持されているので,同様に水素イオンの供給路として機能することが期待される.ビリベルジン–鉄錯体への変化が終わると,新たにポルフィリン環に導入された酸素原子が,ベルドヘムに含まれていた酸素原子と衝突するので,ポルフィリン環にゆがみが生じる.その結果,ピロール環と遠位側へリックスが衝突すると予想される.実際にビリベルジン–鉄錯体複合体では,ヘム複合体やベルドヘム複合体と比べて,遠位側へリックス中の水素結合パターンに変化が生じ,もともと折れ曲がっていた遠位側へリックスの折れ曲がり角度が変化することによって,ヘムポケットが広がっていた(図4b).ビリベルジン–鉄錯体中の鉄原子が還元され二価となり,溶解度が向上すれば,鉄原子はビリベルジンから解離する.この際先述の構造変化によって,ヘムポケットがすでに広がっているので,ビリベルジンも容易に解離すると予想される.
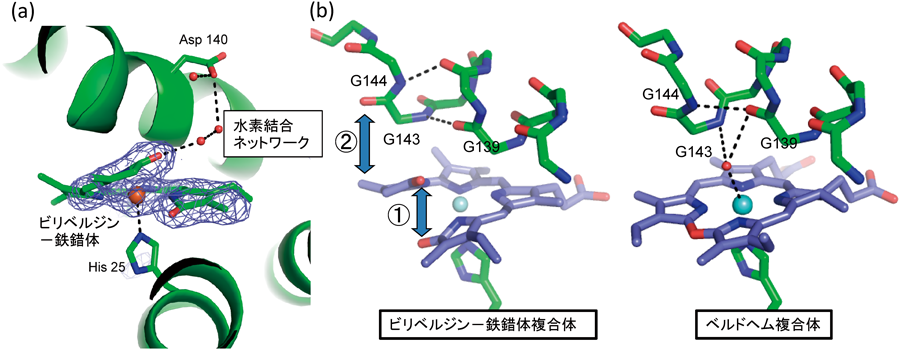
(a)ビリベルジン–鉄錯体周辺の立体構造.網はビリベルジン–鉄錯体をモデルから除いて計算した差電子密度(オミットマップ).A環のラクタム酸素原子とAsp140は水素結合ネットワークで結ばれている.(b)ビリベルジン–鉄錯体複合体とベルドヘム複合体の比較.ポルフィリン環が開裂することによって,ビリベルジン中の酸素原子(赤い球)間に立体障害が生じ,ポルフィリン環がゆがむ(①).その結果,Gly144とA環が衝突し(②),遠位側へリックスの構造変化が生じる.ベルドヘム複合体では,Gly139とGly144が水素結合していた(πヘリックスのコンホメーション)が,ビリベルジン–鉄錯体複合体では,Gly139とGly143, Asp140とGly144(αへリックスのコンホメーション)が水素結合するように構造変化した.
我々はHO-1単体のX線結晶構造も決定しており,そのX線結晶構造中では,近位側へリックスが揺らぎによって,構造決定できない状態であった15).サントリー生物有機科学研究所の菅瀬謙治(現:京都大学)らとの共同研究によるラットHO-1を使ったNMRによる揺らぎの解析からは,HO-1単体でも近位側へリックスのへリックス構造は維持されているが,ヘムポケット周辺(遠位側へリックスと近位側へリックス)とそこから遠く離れたCDループに揺らぎがみられ,この揺らぎはヘムのアナログである亜鉛ポルフィリン錯体を結合させると消失するという結果が得られた(図5)42, 43).すなわち,HO-1単体ではヘムポケットはopenとcloseの間で揺らいでいる状態にあり,ヘムが結合するとcloseへ,反応がビリベルジン–鉄錯体複合体まで至るとopenとなる.さらに,ビリベルジンが解離すると,また揺らいだ状態になるという基質の結合解離と酵素の揺らぎの関係性が描けた.
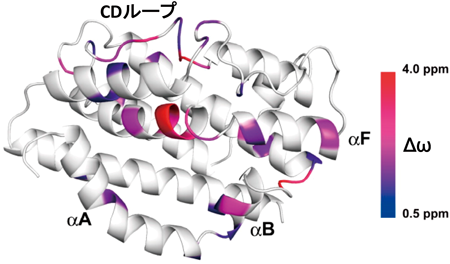
R2 dispersion法75)によって推定した主要コンホメーションとマイナーコンホメーションの化学シフト差(Δω)の大小によって,HO-1の結晶構造(αAはヘム複合体の構造からモデリングしたもの)を色づけた.Δωが大きいほど揺らぎが大きいことを示す.αAは近位側へリックス,αFは遠位側へリックスを示す.文献42より許可を得て転載.Copyright (2015) American Chemical Society.
なお,これらの揺らいでいる箇所(ヘムポケットとCDループ)は,後に示すCPRの結合部位とよく一致する44).CPRとHO-1の相互作用解析結果は,HO-1単体ではCPRと相互作用できないことを示しており,これはHO-1単体での揺らぎに関係すると考えられる.すなわち,この構造揺らぎは単純に誘導適合的な酵素と基質の認識というだけではなくて,CPRとの相互作用も制御する玄妙な機構といえる.
HOはその反応過程で一酸化炭素を放出する.生体内では,この一酸化炭素の大部分は赤血球中のヘモグロビンに結合し,総ヘモグロビンの0.8%程度には一酸化炭素が結合している.外界からの一酸化炭素の摂取が多くなりすぎると,より多くのヘモグロビンに一酸化炭素が結合し,酸素を輸送できなくなる,いわゆる一酸化炭素中毒となる.このように,通常ヘムタンパク質において,一酸化炭素は酸素よりも強くヘム鉄に結合する.ヘモグロビンの場合,それぞれの解離定数を比較すると,一酸化炭素の方が酸素よりも100倍程度強く結合する.しかし,HOでは反応中に生じる一酸化炭素が酵素反応を阻害しないように,ヘム–HO複合体に対する酸素と一酸化炭素の親和性は同程度である45).
HOにおける酸素と一酸化炭素の厳密な識別機構を明らかにするために,筆者らは一酸化炭素が還元型ヘムに結合したHO-1のX線結晶構造を決定し,ヘムに酸素が結合した場合や何も結合していない場合(還元型ヘム複合体)と比較した17).用いた結晶は,酸化型ヘム複合体を結晶化したのちにハイドロサルファイト(亜ジチオン酸ナトリウム)で還元することによって還元型ヘム複合体を調製し,さらに酸素や一酸化炭素を加えることで,酸素結合型や一酸化炭素結合型を調製した.すなわち,もともとの結晶化条件(pHや塩濃度)は同一であり,結晶中のタンパク質分子の充填のされ方も同一(結晶が同型)であるので,軸配位子の違いによる立体構造のわずかな差異を捉えることが可能であった.
酸素と一酸化炭素の親和性が同程度になる要因は二つあり,一つは酸素の親和性増大,もう一つは一酸化炭素の親和性減少である.酸素の親和性増大はHO以外に,ヘモグロビンやミオグロビンでもとられている戦略である.有機溶媒中のヘムは一酸化炭素を酸素の1万倍以上強く結合するが,ヘモグロビンやミオグロビンでは,酸素の親和性が増大している結果,親和性の比は数十倍から数百倍に抑えられている46).HOでも酸素分子の片方の酸素原子は水分子と,もう片方の酸素原子はGly143のアミド基と水素結合して安定化されることによって,酸素の親和性が増大していることがわかった(図3a).
HOで特別にみられたこととして,一酸化炭素が結合した場合にのみ,遠位側へリックスとヘムがそれぞれ逆方向に移動し,一酸化炭素が結合できる空間を作っていることがあげられる(図6a).酸素分子は酸素原子どうしが二重結合で,一酸化炭素は酸素原子と炭素原子が三重結合で結合しているので,それぞれの分子軌道(酸素分子はsp2混成軌道,一酸化炭素はsp混成軌道)を考えると,ヘム鉄と酸素分子は120°の,ヘム鉄と一酸化炭素分子は180°の結合角で結合するのが理想的である.この結合角度とHOの立体構造を考えると,酸素分子が結合する場合には遠位側へリックスと酸素分子は衝突しないが,一酸化炭素が結合する場合には遠位側へリックスと一酸化炭素が衝突することになる.実際には,その衝突によってへリックスとヘムが移動したと考えられる.同様の立体構造変化はシアン(180°の結合角が安定)がヘム鉄に結合した場合にもみられ,アジドや一酸化窒素(120°の結合角が安定)が結合した場合にはみられなかった.これは,ヘモグロビンやミオグロビンと比較してヘム鉄の遠位側の空間が非常に狭くなっている特徴を反映したものであり,HO特有の酸素と一酸化炭素の識別機構といえる.
この立体構造変化は可逆的である(図6b).シカゴ大学のKeith Moffatらとの共同研究により100 Kまで冷却した結晶中のヘム鉄と一酸化炭素の結合をレーザー光の照射で切断し,その後の構造変化を解析したところ,移動していたへリックスとヘムが元の位置に戻るようすがX線結晶構造解析で観察された18).おそらく,レーザー光照射により,結晶が160 K付近まで温められた結果,構造変化が可能になったと考えている.また,同様の光照射実験(T=~30 K, 100 K)から,ヘムポケットから一酸化炭素が放出されていく際に一時的にトラップされる部位もいくつか発見された18, 47).ミオグロビンの場合,ヘムポケットの遠位側から近位側に一酸化炭素が移動するようすがみられるが48, 49),HOの場合は,遠位側のみにトラップされていた.HO反応では,ベルドヘムと同時に一酸化炭素が生成するが,この一酸化炭素はこれらのトラップ部位を通って,HOの外側へと遊離していくと予想される.
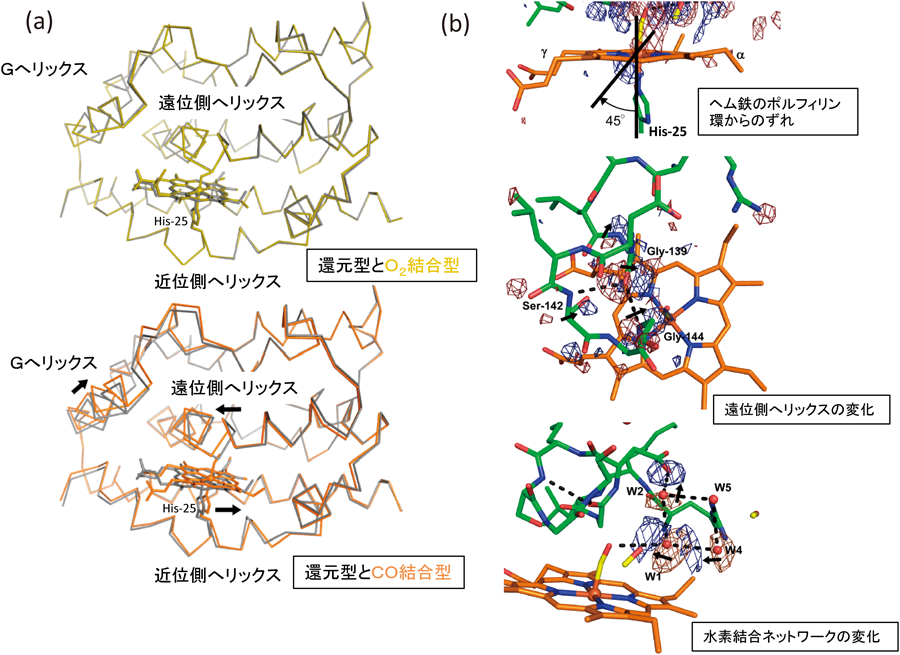
(a)還元型ヘム複合体(5配位型,灰色)とO2結合型(6配位型,黄色)またはCO結合型(6配位型,橙色)との重ね合わせ.O2結合型との重ね合わせでは,ほとんど構造の違いはみられないが,CO結合型との重ね合わせでは,矢印の位置にずれが生じている.(b)一酸化炭素の光解離前後の構造変化.網はレーザー光照射前後の差電子密度(赤は負の,青は正の電子密度).赤と青の差電子密度が原子モデル周辺にみられた場合は,一酸化炭素が解離したことによって,原子が赤から青の方向に移動したことを示す.矢印で示したように,ヘム鉄のHis25方向への移動,遠位側へリックスの移動[(a)とは逆方向],水素結合ネットワークを構成する水分子(W1, W2, W4)の移動が観察された.これらの変化は結晶中で160 K(約−113°C)以下で起きたものである.文献18より許可を得て転載.Copyright (2012) American Chemical Society.
さらに,大阪大学の水谷泰久らとの共同研究による溶液を使った室温での時間分解共鳴ラマン分光法による解析では,ヘム鉄の近位側軸配位子の結合長やヘムプロピオン酸のコンホメーション変化に起因すると考えられるスペクトル変化が一酸化炭素の解離からns~µsの時間スケールで起きていることがわかった50).ミオグロビンなど他のヘムタンパク質でも,一酸化炭素の光解離後の構造ダイナミクスを調べるために,時間分解共鳴ラマン分光法による解析が行われている51)が,その結果と比べると比較的遅いといえる.また,軸配位子の結合長の変化にも特徴(Fe-His伸縮振動数の増加)がみられた.一般的に一酸化炭素が光解離すると,ヘムが6配位構造から5配位構造へと変化するので,ヘム鉄がポルフィリン環からずれ込む.その結果,いったん軸配位子との結合長が短くなった後,結合長が緩和していく過程が観察される(Fe-His伸縮振動数の減少).この過程に加えて,HOの場合,ヘム全体の並進による影響が混じりあう(結晶中では図6bのように観察される)ので,特徴的な変化が観察されたと考えている.すなわち,溶液状態でも結晶でみられたHOの構造変化を反映していると考えられる影響がみられた.
HOは酵素反応を完了するために7個の電子を必要とする.これらの電子を哺乳類のHOはCPRから,植物などの光合成生物のHOはフェレドキシンから受け取る.これら以外の細菌由来HOがどの分子から電子を受け取るのかは不明である.CPRはフラビンモノヌクレオチド(FMN)とフラビンアデニンジヌクレオチド(FAD)を1分子ずつ補酵素として結合した膜結合型フラビン酵素であり,フェレドキシンは鉄硫黄クラスターを補酵素とする可溶性タンパク質である.
CPRはNADPH→FAD→FMN→還元相手(ヘム)へと電子を供給する.CPRの立体構造は1990年代にウィスコンシン医科大学のJung-Ja Kimらによって,ラット由来CPRの構造が報告されている52).この立体構造中でNADPH, FAD, FMNは互いに近接しており,CPR内で電子移動を行うためには非常に都合のよい立体構造であった.しかし,FMNがCPR内部に埋もれているため,HOやシトクロムP450に電子移動を行う場合には,都合の悪い立体構造と考えられていた.実際に,変異CPRとシトクロムP450を使った相互作用解析結果から得られた両者の相互作用に関与するアミノ酸残基とCPRの立体構造はつじつまが合わないものであった53).この結果はHOやシトクロムP450とCPRが相互作用する場合には,CPRに大きな構造変化が生じることを示唆するものであった.
その後,2000年代後半に,従来報告されていたCPRの立体構造とは別のコンホメーションの立体構造が報告された.一つは酵母CPRとヒトCPRのキメラタンパク質の結晶構造54),もう一つはラットCPRのFMN結合ドメインとFAD結合ドメインの間のループに存在するアミノ酸残基を4残基短縮した変異CPR(ΔTGEE)の結晶構造である55).従来の構造(close型)では,FMNがCPR内部に埋もれていたが,これらの新しい構造(open型)ではFMN結合ドメインが,FAD結合ドメインから離れて,自由に動けるような立体構造であった.さらに,溶液状態で大まかな立体構造を得ることができるX線小角散乱法による解析56, 57)やフラビン間に生じるFRETを解析した結果58)から,塩濃度や酸化還元状態を変化させることで,CPRがclose型とopen型を行き来することが示唆された.
筆者らはΔTGEEとヘム–HO-1複合体が強く相互作用することを,ゲルろ過クロマトグラフィーや表面プラズモン共鳴法による相互作用解析から見いだし,ゲルろ過クロマトグラフィーで得られたΔTGEEとヘム–HO-1の複合体画分を結晶化した.この結晶は4.3 Å分解能と低分解能までしか回折しなかったが,ΔTGEEとヘム–HO-1の複合体の結晶構造解析に成功した(図7a)44).ΔTGEE自身は非常にフレキシブルな構造変化が可能なため,結晶化によって本来の複合体の結合様式とは異なるものが得られている可能性があった.そこで,結晶構造が溶液状態でも同様であるかどうかX線小角散乱法による解析を行ったところ,溶液状態でも同様の複合体であることが示された44).また,ΔTGEEは変異CPRであるので,本来のCPRとは別の結合様式でHOと結合している可能性もあった.そこで,4残基短縮した部分を元のアミノ酸配列に戻し,ΔTGEEとヘム–HO-1の複合体の結晶構造中で近傍に存在していたΔTGEEとHO-1のアミノ酸残基の組み合わせを両方ともシステインに置換した変異CPRと変異HO-1を調製した.これらの間で架橋実験を行ったところ,変異に由来するジスルフィド結合の生成が認められた(元のラットHO-1にはシステイン残基は含まれていない)44).また,ΔTGEEもHO-1も本来は膜結合タンパク質であるが,結晶化にあたって,膜結合部位(ΔTGEEではN末端側,HO-1ではC末端側)を除去して可溶化した酵素を用いている.それにも関わらずΔTGEEのN末端側,HO-1のC末端側は結晶構造中で同じ方向を向いており,細胞内のタンパク質の配置を考えても矛盾しない構造であった.したがって,ΔTGEEとヘム–HO-1の複合体の結晶構造はCPRとヘム–HO-1の複合体の結合様式を忠実に反映しているといえる.
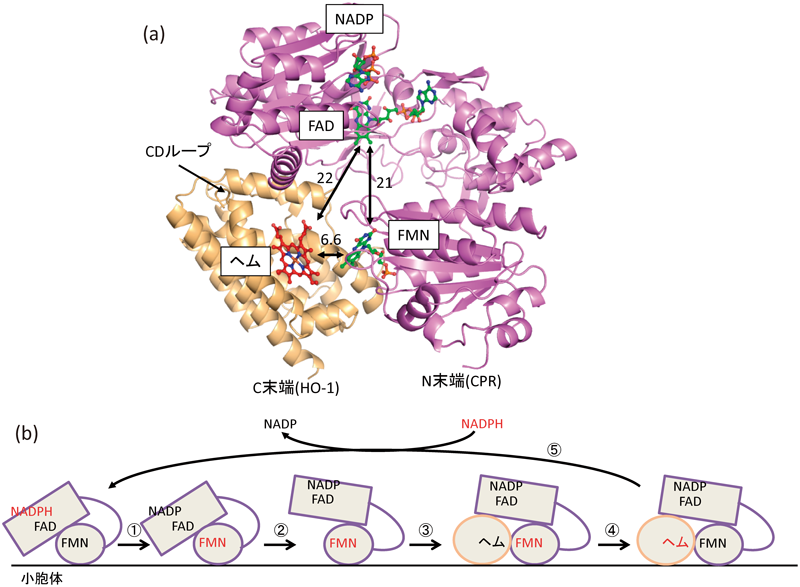
(a)CPR (ΔTGEE)–ヘム–HO-1複合体の立体構造.紫はCPR, カーキ色はHOを示す.数字は補欠分子族間の最短距離をÅ単位で示した.(b)CPRからHOへの電子移動モデル.CPRもHOも細胞内では小胞体膜上に存在する.赤色はその補欠分子族が還元型であることを示す.①close型CPRにNADPHが結合して,分子内電子移動が生じる.②FMNが還元されると,open型が安定化される.③open型CPRにヘム–HO複合体が結合する(結晶構造).④還元型FMNからヘムに分子間電子移動が生じる.⑤FMNが酸化され,close型が安定化され,HOはCPRから解離する.
ΔTGEEとヘム–HO-1の複合体の結晶構造中で,FMNはヘムと最短で6.6 Åと近接しており,直接電子移動可能な距離にあったが,FADとFMNは20 Å以上離れており,直接電子移動が可能とは考えられない位置関係であった.実際にΔTGEEとヘム–HO-1間の電子移動速度を測定したところ,野生型CPRを使った場合の約1/360の速度であり,この立体構造のままでは効率よく電子移動が行えないことがわかった44).すなわち,CPRからヘム–HO-1への電子移動では,CPRのclose型とopen型の調節された構造変化が必要であることが示唆された.これらの結果から推定した電子移動モデルを図7bに示す.今後,この電子移動モデルの生化学的な検証と結晶構造の分解能向上を目指していきたいと考えている.
HOによって生成されたビリベルジンは,動物ではNADH/NADPH依存的なビリベルジン還元酵素によって,ビリルビンへと転換される.一方,植物などの光合成生物では,動物のビリベルジン還元酵素とはまったく相同性のないFDBRによって,光合成色素(フィコビリン)や光受容色素(フィトクロモビリン)が合成される11, 12).筆者らはFDBRの一種であるPcyAについて,立体構造決定を行ったので,それについて紹介する.
PcyAはビリベルジンを基質として二段階の還元反応(図8a)によって,フィコシアノビリンを合成する酵素である.筆者らはシアノバクテリアの一種であるSynechocystis sp. PCC6803由来PcyAを大腸菌を使った組換え酵素として発現させ,PcyAとビリベルジンの複合体のX線結晶構造解析に成功した(図8b)59).ビリベルジンは基質であるが,フェレドキシンがない状態では,酵素と安定な複合体を形成する.PcyAは類縁酵素の立体構造がまったく報告されていなかったので,重原子置換体結晶を作製し,新規に立体構造決定を行った.PcyAの全体構造はα/β/αサンドイッチ構造で,中央のβシートとC末端側のαへリックスの間にビリベルジンが結合していた.予想していなかったことに,全体構造はポルフィリン合成系で機能する酸素依存性コプロポルフィリノーゲンオキシダーゼとよく似ていた60).両酵素間にアミノ酸配列レベルでの相同性はみられない.また,後にわかったことだが,アミノ酸配列レベルでの相同性がかなり低いRCCRという酵素(クロロフィル分解系で機能する酵素)61)とも立体構造はよく似ていた.これらの酵素はすべてテトラピロール類縁の化合物を基質とするものである.
ビリベルジンのプロピオン酸側鎖はリシンやアルギニンといった塩基性残基と相互作用していた(図8c).PcyA表面の電荷分布をみると,プロピオン酸側鎖が相互作用している部位が特徴的に正に帯電していたので,酸性タンパク質であるフェレドキシンはビリベルジンのプロピオン酸側鎖近傍に結合し,プロピオン酸側鎖を介して電子移動を行うことが予想された.ビリベルジンのテトラピロール部分では,四つのピロール環の中心付近にAsp105が存在し,ピロール環の窒素原子と水素結合していた.このAsp105の側鎖は二つのコンホメーションをとっており,そのうちの一つはピロール環の一つ(D環)のラクタム酸素原子と水素結合していた(図8c).このD環に結合しているビニル側鎖は,PcyAによって最初に還元される部位である.D環ビニル側鎖の近傍にはGlu76が存在し,この残基は最初の還元反応に関与する触媒残基と推定された.また,D環とA環のラクタム酸素原子の近傍にはHis88側鎖が存在し,His88とこれらのラクタム酸素原子は水素結合していた(図8c).後の変異実験から,これらの三つのアミノ酸残基はいずれも触媒活性に関与することが示唆された62, 63).
その後,筆者らはビリベルジンを結合していないPcyA単体64)や反応中間体の181,182-ジヒドロビリベルジンとの複合体65)のX線結晶構造も決定した.PcyA単体の結晶構造中では,His88とAsp105が水素結合しており,ビリベルジンが結合することによってその水素結合パターンが変化することが示された.現在考えられているPcyAの反応モデルでは,中性のビリベルジンはPcyAに結合することによって塩基性に変化することが示唆されている.PcyAにビリベルジンが結合した際の構造変化は,塩基性への変化に必要な水素イオンの供給にAsp105が関与していることを示している.181,182-ジヒドロビリベルジンとの複合体構造は,ビリベルジン複合体とそれほど変わらない構造であったが,18位のビニル基がエチル基に変化したことによって,その近傍にあったGlu76側鎖のコンホメーションが変化した.ただ,ビリベルジン複合体のGlu76側鎖のコンホメーションには,X線照射による影響がみられることが後にわかった66)ので,181,182-ジヒドロビリベルジン複合体でみられたGlu76のコンホメーション変化にはさらなる検討が必要である.
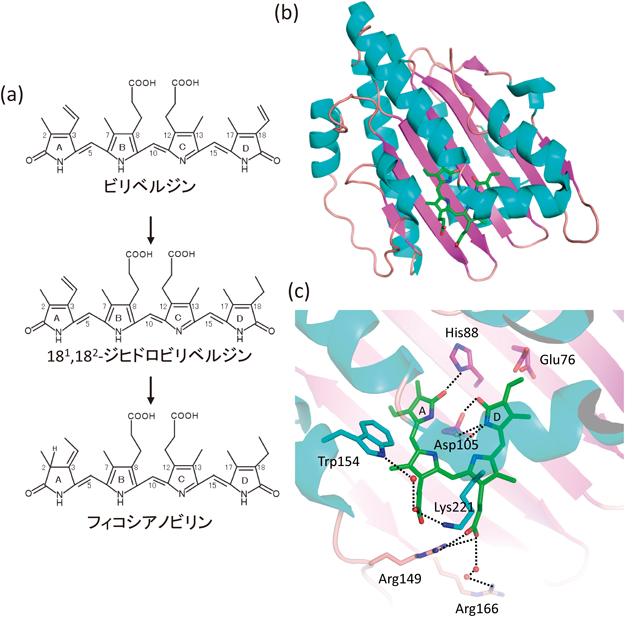
(a)PcyAの反応スキーム.一段階目の反応では,D環のビニル基が還元され,二段階目の反応では,A環が還元される.(b)PcyA–ビリベルジン複合体の立体構造.二次構造によって色分けしている.(c)ビリベルジン周辺の構造.点線は水素結合または塩橋を示す.プロピオン酸側鎖やピロール環との水素結合によって,結合が安定化されている.
筆者ら以外にも,カリフォルニア大学のClark Lagariasやルール大学ボーフムのNicole Frankenberg-Dinkel, Academia SinicaのShi-Long Tuらの研究グループはPcyA–ビリベルジン複合体を一電子還元させた状態(ラジカル状態)の結晶構造や分光学的解析67, 68),PcyAとは別のFDBR(ビリベルジンからフィコエリスロビリンを合成する酵素群)の結晶構造解析結果69, 70)を報告し,基質認識や反応部位の特異性について議論している.さらに,筆者らは茨城大学の海野昌喜らとの共同研究でPcyA–ビリベルジン複合体の中性子結晶解析にも成功しており,水素原子まで含めた立体構造が明らかとなった66).詳細については,昨年の本誌「みにれびゅう」に掲載しているので,ご一読いただきたい71).
哺乳類のビリベルジン還元酵素については,理化学研究所の城宜嗣と山形大学の吉田匡らの研究グループやロチェスター大学のMahin Mainesらによって,ビリベルジン還元酵素単体やNAD, NADPが結合した状態の結晶構造が報告されている72, 73).しかし,ビリベルジンが結合した状態の結晶構造は報告されておらず,ビリベルジンの結合位置や反応に関与する触媒残基などは推定されているのみである.筆者らはSynechocystis sp. PCC6803由来ビリベルジン還元酵素ホモログの結晶化に成功している74).現在,ビリベルジンとNADPが結合した状態の結晶も得られており,その立体構造から,ビリベルジン還元酵素の還元反応に関する構造基盤が得られることを期待している.
これらの研究は,18年前に筆者が大阪大学大学院理学研究科生物科学専攻の福山恵一先生(現:大阪大学大学院工学研究科)の研究室に配属されたころから,久留米大学医学部の野口正人先生(現:帝京大学福岡医療技術学部)との共同研究として始めたものです.当時の福山研究室および野口研究室の皆様,ならびに数多くの共同研究者の方々に感謝申し上げます.また,実験結果のほとんどは放射光実験によって得られたものです.実験を行わせていただいた大型放射光施設SPring-8, Advanced Photon Source(米国イリノイ州),佐賀県立九州シンクロトロン光研究センターの関係各位に感謝いたします.また,これらの研究は科研費および石橋学術振興基金助成金の支援を受けており,感謝いたします.
最後に,現所属の久留米大学医学部医化学講座(山本健教授)の皆様には,研究全般にわたって,ご指導ご協力いただいております.深く感謝いたします.
1) Tenhunen, R., Marver, H.S., & Schmid, R. (1969) J. Biol. Chem., 244, 6388–6394.
2) Ortiz de Montellano, P.R. (2000) Curr. Opin. Chem. Biol., 4, 221–227.
3) Kikuchi, G., Yoshida, T., & Noguchi, M. (2005) Biochem. Biophys. Res. Commun., 338, 558–567.
4) Noguchi, M., Yoshida, T., & Kikuchi, G. (1979) J. Biochem., 86, 833–848.
5) Stocker, R., Yamamoto, Y., McDonagh, A.F., Glazer, A.N., & Ames, B.N. (1987) Science, 235, 1043–1046.
6) Alam, J., Stewart, D., Touchard, C., Boinapally, S., Choi, A.M., & Cook, J.L. (1999) J. Biol. Chem., 274, 26071–26078.
7) Ryter, S.W., Otterbein, L.E., Morse, D., & Choi, A.M. (2002) Mol. Cell. Biochem., 234–235, 249–263.
8) Morikawa, T., Kajimura, M., Nakamura, T., Hishiki, T., Nakanishi, T., Yukutake, Y., Nagahata, Y., Ishikawa, M., Hattori, K., Takenouchi, T., Takahashi, T., Ishii, I., Matsubara, K., Kabe, Y., Uchiyama, S., Nagata, E., Gadalla, M.M., Snyder, S.H., & Suematsu, M. (2012) Proc. Natl. Acad. Sci. USA, 109, 1293–1298.
9) Shintani, T., Iwabuchi, T., Soga, T., Kato, Y., Yamamoto, T., Takano, N., Hishiki, T., Ueno, Y., Ikeda, S., Sakuragawa, T., Ishikawa, K., Goda, N., Kitagawa, Y., Kajimura, M., Matsumoto, K., & Suematsu, M. (2009) Hepatology, 49, 141–150.
10) Dioum, E.M., Rutter, J., Tuckerman, J.R., Gonzalez, G., Gilles-Gonzalez, M.A., & McKnight, S.L. (2002) Science, 298, 2385–2387.
11) Frankenberg, N., Mukougawa, K., Kohchi, T., & Lagarias, J.C. (2001) Plant Cell, 13, 965–978.
12) Kohchi, T., Mukougawa, K., Frankenberg, N., Masuda, M., Yokota, A., & Lagarias, J.C. (2001) Plant Cell, 13, 425–436.
13) Ikeuchi, M. & Ishizuka, T. (2008) Photochem. Photobiol. Sci., 7, 1159–1167.
14) Wagner, J.R., Brunzelle, J.S., Forest, K.T., & Vierstra, R.D. (2005) Nature, 438, 325–331.
15) Sugishima, M., Sakamoto, H., Kakuta, Y., Omata, Y., Hayashi, S., Noguchi, M., & Fukuyama, K. (2002) Biochemistry, 41, 7293–7300.
16) Sugishima, M., Omata, Y., Kakuta, Y., Sakamoto, H., Noguchi, M., & Fukuyama, K. (2000) FEBS Lett., 471, 61–66.
17) Sugishima, M., Sakamoto, H., Noguchi, M., & Fukuyama, K. (2003) Biochemistry, 42, 9898–9905.
18) Sugishima, M., Moffat, K., & Noguchi, M. (2012) Biochemistry, 51, 8554–8562.
19) Sato, H., Sugishima, M., Sakamoto, H., Higashimoto, Y., Shimokawa, C., Fukuyama, K., Palmer, G., & Noguchi, M. (2009) Biochem. J., 419, 339–345.
20) Sugishima, M., Sakamoto, H., Higashimoto, Y., Noguchi, M., & Fukuyama, K. (2003) J. Biol. Chem., 278, 32352–32358.
21) Schuller, D.J., Wilks, A., Ortiz de Montellano, P.R., & Poulos, T.L. (1999) Nat. Struct. Biol., 6, 860–867.
22) Fujii, H., Zhang, X., & Yoshida, T. (2004) J. Am. Chem. Soc., 126, 4466–4467.
23) Caignan, G.A., Deshmukh, R., Wilks, A., Zeng, Y., Huang, H.W., Moenne-Loccoz, P., Bunce, R.A., Eastman, M.A., & Rivera, M. (2002) J. Am. Chem. Soc., 124, 14879–14892.
24) Wilks, A., Torpey, J., & Ortiz de Montellano, P.R. (1994) J. Biol. Chem., 269, 29553–29556.
25) Sugishima, M., Sakamoto, H., Higashimoto, Y., Omata, Y., Hayashi, S., Noguchi, M., & Fukuyama, K. (2002) J. Biol. Chem., 277, 45086–45090.
26) Unno, M., Matsui, T., Chu, G.C., Couture, M., Yoshida, T., Rousseau, D.L., Olson, J.S., & Ikeda-Saito, M. (2004) J. Biol. Chem., 279, 21055–21061.
27) Fujii, H., Zhang, X., Tomita, T., Ikeda-Saito, M., & Yoshida, T. (2001) J. Am. Chem. Soc., 123, 6475–6484.
28) Matsui, T., Furukawa, M., Unno, M., Tomita, T., & Ikeda-Saito, M. (2005) J. Biol. Chem., 280, 2981–2989.
29) Chen, H., Moreau, Y., Derat, E., & Shaik, S. (2008) J. Am. Chem. Soc., 130, 1953–1965.
30) Kamachi, T., Shestakov, A.F., & Yoshizawa, K. (2004) J. Am. Chem. Soc., 126, 3672–3673.
31) Matera, K.M., Takahashi, S., Fujii, H., Zhou, H., Ishikawa, K., Yoshimura, T., Rousseau, D.L., Yoshida, T., & Ikeda-Saito, M. (1996) J. Biol. Chem., 271, 6618–6624.
32) Migita, C.T., Fujii, H., Mansfield Matera, K., Takahashi, S., Zhou, H., & Yoshida, T. (1999) Biochim. Biophys. Acta, 1432, 203–213.
33) Sakamoto, H., Omata, Y., Palmer, G., & Noguchi, M. (1999) J. Biol. Chem., 274, 18196–18200.
34) Sakamoto, H., Takahashi, K., Higashimoto, Y., Harada, S., Palmer, G., & Noguchi, M. (2005) Biochem. Biophys. Res. Commun., 338, 578–583.
35) Zhang, X., Fujii, H., Mansfield Matera, K., Migita, C.T., Sun, D., Sato, M., Ikeda-Saito, M., & Yoshida, T. (2003) Biochemistry, 42, 7418–7426.
36) Lad, L., Ortiz de Montellano, P.R., & Poulos, T.L. (2004) J. Inorg. Biochem., 98, 1686–1695.
37) Takahashi, S., Mansfield Matera, K., Fujii, H., Zhou, H., Ishikawa, K., Yoshida, T., Ikeda-Saito, M., & Rousseau, D.L. (1997) Biochemistry, 36, 1402–1410.
38) Damaso, C.O., Bunce, R.A., Barybin, M.V., Wilks, A., & Rivera, M. (2005) J. Am. Chem. Soc., 127, 17582–17583.
39) Unno, M., Matsui, T., & Ikeda-Saito, M. (2012) J. Inorg. Biochem., 113, 102–109.
40) Sakamoto, H., Omata, Y., Adachi, Y., Palmer, G., & Noguchi, M. (2000) J. Inorg. Biochem., 82, 113–121.
41) Lai, W., Chen, H., Matsui, T., Omori, K., Unno, M., Ikeda-Saito, M., & Shaik, S. (2010) J. Am. Chem. Soc., 132, 12960–12970.
42) Harada, E., Sugishima, M., Harada, J., Fukuyama, K., & Sugase, K. (2015) Biochemistry, 54, 340–348.
43) Harada, E., Sugishima, M., Harada, J., Noguchi, M., Fukuyama, K., & Sugase, K. (2015) Biomol. NMR Assign., 9, 197–200.
44) Sugishima, M., Sato, H., Higashimoto, Y., Harada, J., Wada, K., Fukuyama, K., & Noguchi, M. (2014) Proc. Natl. Acad. Sci. USA, 111, 2524–2529.
45) Migita, C.T., Mansfield Matera, K., Ikeda-Saito, M., Olson, J.S., Fujii, H., Yoshimura, T., Zhou, H., & Yoshida, T. (1998) J. Biol. Chem., 273, 945–949.
46) Springer, B.A., Sligar, S.G., Olson, J.S., & Phillips, G.N. Jr. (1994) Chem. Rev., 94, 699–714.
47) Sugishima, M., Sakamoto, H., Noguchi, M., & Fukuyama, K. (2004) J. Mol. Biol., 341, 7–13.
48) Chu, K., Vojtchovský, J., McMahon, B.H., Sweet, R.M., Berendzen, J., & Schlichting, I. (2000) Nature, 403, 921–923.
49) Tomita, A., Sato, T., Ichiyanagi, K., Nozawa, S., Ichikawa, H., Chollet, M., Kawai, F., Park, S.Y., Tsuduki, T., Yamato, T., Koshihara, S.Y., & Adachi, S. (2009) Proc. Natl. Acad. Sci. USA, 106, 2612–2616.
50) Yamaoka, M., Sugishima, M., Noguchi, M., Fukuyama, K., & Mizutani, Y. (2011) J. Raman Spectrosc., 42, 910–916.
51) Mizutani, Y. & Kitagawa, T. (2001) Chem. Rec., 1, 258–275.
52) Wang, M., Roberts, D.L., Paschke, R., Shea, T.M., Masters, B.S., & Kim, J.J. (1997) Proc. Natl. Acad. Sci. USA, 94, 8411–8416.
53) Kasper, C.B. (1995) J. Biol. Chem., 270, 27475–27480.
54) Aigrain, L., Pompon, D., Morera, S., & Truan, G. (2009) EMBO Rep., 10, 742–747.
55) Hamdane, D., Xia, C., Im, S.C., Zhang, H., Kim, J.J., & Waskell, L. (2009) J. Biol. Chem., 284, 11374–11384.
56) Ellis, J., Gutierrez, A., Barsukov, I.L., Huang, W.C., Grossmann, J.G., & Roberts, G.C. (2009) J. Biol. Chem., 284, 36628–36637.
57) Huang, W.C., Ellis, J., Moody, P.C., Raven, E.L., & Roberts, G.C. (2013) Structure, 21, 1581–1589.
58) Pudney, C.R., Khara, B., Johannissen, L.O., & Scrutton, N.S. (2011) PLoS Biol., 9, e1001222.
59) Hagiwara, Y., Sugishima, M., Takahashi, Y., & Fukuyama, K. (2006) Proc. Natl. Acad. Sci. USA, 103, 27–32.
60) Phillips, J.D., Whitby, F.G., Warby, C.A., Labbe, P., Yang, C., Pflugrath, J.W., Ferrara, J.D., Robinson, H., Kushner, J.P., & Hill, C.P. (2004) J. Biol. Chem., 279, 38960–38968.
61) Sugishima, M., Kitamori, Y., Noguchi, M., Kohchi, T., & Fukuyama, K. (2009) J. Mol. Biol., 389, 376–387.
62) Tu, S.L., Sughrue, W., Britt, R.D., & Lagarias, J.C. (2006) J. Biol. Chem., 281, 3127–3136.
63) Tu, S.L., Rockwell, N.C., Lagarias, J.C., & Fisher, A.J. (2007) Biochemistry, 46, 1484–1494.
64) Hagiwara, Y., Sugishima, M., Takahashi, Y., & Fukuyama, K. (2006) FEBS Lett., 580, 3823–3828.
65) Hagiwara, Y., Sugishima, M., Khawn, H., Kinoshita, H., Inomata, K., Shang, L., Lagarias, J.C., Takahashi, Y., & Fukuyama, K. (2010) J. Biol. Chem., 285, 1000–1007.
66) Unno, M., Ishikawa-Suto, K., Kusaka, K., Tamada, T., Hagiwara, Y., Sugishima, M., Wada, K., Yamada, T., Tomoyori, K., Hosoya, T., Tanaka, I., Niimura, N., Kuroki, R., Inaka, K., Ishihara, M., & Fukuyama, K. (2015) J. Am. Chem. Soc., 137, 5452–5460.
67) Stoll, S., Gunn, A., Brynda, M., Sughrue, W., Kohler, A.C., Ozarowski, A., Fisher, A.J., Lagarias, J.C., & Britt, R.D. (2009) J. Am. Chem. Soc., 131, 1986–1995.
68) Kohler, A.C., Gae, D.D., Richley, M.A., Stoll, S., Gunn, A., Lim, S., Martin, S.S., Doukov, T.I., Britt, R.D., Ames, J.B., Lagarias, J.C., & Fisher, A.J. (2010) Biochemistry, 49, 6206–6218.
69) Busch, A.W., Reijerse, E.J., Lubitz, W., Frankenberg-Dinkel, N., & Hofmann, E. (2011) Biochem. J., 439, 257–264.
70) Busch, A.W., Reijerse, E.J., Lubitz, W., Hofmann, E., & Frankenberg-Dinkel, N. (2011) Biochem. J., 433, 469–476.
71) 福山恵一,和田啓,杉島正一,海野昌喜(2015)生化学,87, 753–757.
72) Kikuchi, A., Park, S.Y., Miyatake, H., Sun, D., Sato, M., Yoshida, T., & Shiro, Y. (2001) Nat. Struct. Biol., 8, 221–225.
73) Whitby, F.G., Phillips, J.D., Hill, C.P., McCoubrey, W., & Maines, M.D. (2002) J. Mol. Biol., 319, 1199–1210.
74) Watanabe, A., Hirata, K., Hagiwara, Y., Yutani, Y., Sugishima, M., Yamamoto, M., Fukuyama, K., & Wada, K. (2011) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 67, 313–317.
75) 菅瀬謙治(2008)生化学,80, 754–758.

久留米大学医学部准教授.博士(理学).
1976年兵庫県に生る.99年大阪大学理学部生物学科卒業.2004年同大学院理学研究科生物科学専攻博士後期課程修了.同研究科特任研究員,日本学術振興会特別研究員を経て,07年4月より久留米大学医学部助教.10年8月~12年7月シカゴ大学客員研究員を兼任.12年11月より現職.
研究テーマと抱負ポルフィリン代謝に関わるタンパク質や金属タンパク質の構造生物学的研究.これからも継続的な息の長い研究を続けていくと同時に,新規プロジェクトにも取り組んでいきたい.
ウェブサイトhttps://sites.google.com/site/msugishima76/
趣味ドライブ,ハイキング.
本総説は2015年度奨励賞を受賞した.

This page was created on 2016-02-24T19:16:06.803+09:00
This page was last modified on 2016-04-14T16:20:15.714+09:00
このサイトは(株)国際文献社によって運用されています。