低酸素に対する生体反応(低酸素応答)の多くは,転写因子HIFによって制御されていることが知られている1).
HIFは造血,血管新生,炎症,アポトーシス,オートファジーを含め,さまざまなイベントに関与する遺伝子の転写をつかさどっているが,その他にも細胞内エネルギー代謝に関する遺伝子群の転写を制御しており,HIFの活性化は嫌気解糖に関与するトランスポーターや代謝酵素を誘導して細胞のエネルギー代謝をミトコンドリアにおける酸化的リン酸化から細胞質での嫌気解糖へとシフトさせる.しかし,そのHIFもまた酸素濃度センサー分子・PHDによって負に制御されているため,PHDが低酸素時の細胞内エネルギー代謝を制御しているといえる2).
しかしながら,実際には後述するとおり低酸素環境下ではPHDやHIF非依存的なさまざまな低酸素応答が惹起される.また,HIFといっても,その三つのαサブユニット(HIF1α, HIF2α, HIF3α)には,それぞれ発現臓器・転写ターゲット・転写活性に差があるので(HIFといいながらも科学的根拠なしに最初にクローニングされたHIF1αにしか注視していない学会発表や論文が散見されるが),低酸素応答=HIFでもなければHIF=HIF1αでもない.
このようなことを踏まえたうえで,本稿では低酸素環境に対する応答反応のなかで,最もよく知られた酸素濃度センサーの一つであるプロリン水酸化酵素PHDによる細胞内エネルギー代謝制御機構についての筆者らの研究を報告したい.
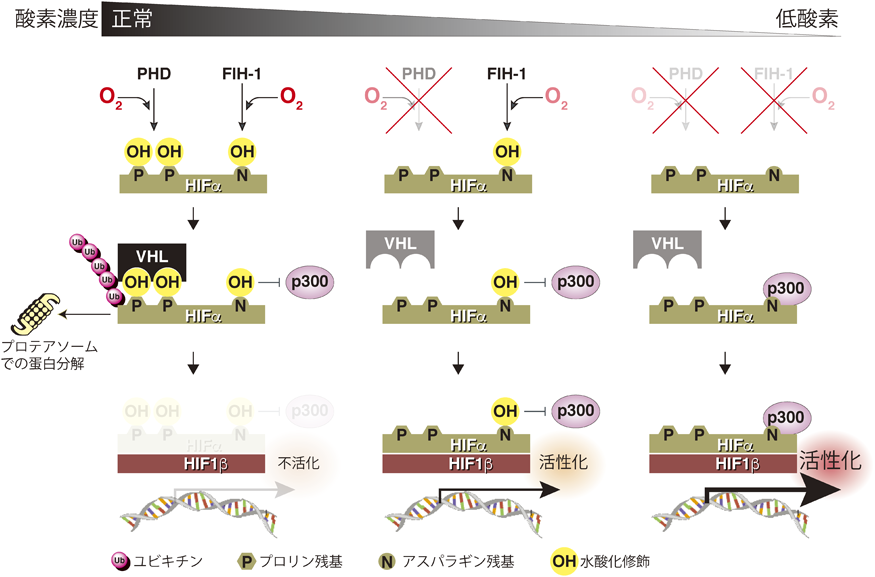
HIFはαとβの二つのサブユニットのヘテロ二量体で形成される転写因子であり,数多くの低酸素関連遺伝子の転写をつかさどる.HIFのβ-サブユニット(HIF1β/ARNT)は恒常的に発現しているのに対して,α-サブユニット(HIFα;HIF1α~3α)は酸素濃度によって発現量が変動する.正常酸素濃度下では,HIFαのN末端側の転写活性化ドメイン(NTAD)内の特定のプロリン残基がプロリン水酸化酵素PHDによって水酸化され,それを指標にHIFαはVHL(von Hippel-Lindau)病の原因遺伝子産物pVHLを含むユビキチンリガーゼ複合体(VBC complex)によってユビキチン化され,プロテアソームでのタンパク質分解へと導かれる(図1).そのため,HIFはHIFα–HIF1βからなるヘテロ二量体を形成できず,HIFを介した低酸素応答は抑制される.一方,低酸素環境においては,その酵素活性に酸素分子を必要とするPHDの活性が低下し,HIFαはpVHL複合体によるユビキチン化を介したタンパク質分解を免れて急速に細胞内に蓄積し,HIF1β/ARNTと結合してヘテロ二量体を形成してゲノムDNA上の特定の配列(HRE:5′-R(A/G)CGT G-3′)を持つ低酸素関連遺伝子群の転写を亢進させ,HIFを介した低酸素応答が活性化する3).
哺乳動物には三つのPHD遺伝子が同定されており,発現臓器や細胞内局在がそれぞれ異なることから,各々の遺伝子には固有の機能があるものと思われる4, 5).in vitroでは三つのPHDともHIFαの特定のプロリン残基を水酸化するが6),in vivoではPHD2が主要なHIFαのプロリン水酸化酵素であり,また発生に必須な分子である7, 8).PHD2は他の2分子と強調しながらHIFαのプロリン残基を水酸化することで,HIFを介した低酸素応答を負に制御している9).
HIFαにはHIF1α, HIF2α, HIF3αの三つの別個の遺伝子から転写されるアイソフォームが同定されており,HIF1αとHIF2αのC末端側の転写活性ドメイン(CTAD)の特定のアスパラギン残基はFIH-1(factor inhibiting HIF1α)によって酸素濃度依存的に水酸化修飾を受ける.このアスパラギン残基の水酸化はヒストンアセチル基転移酵素(HAT)活性を持った転写共役因子であるp300とCTADとの結合を阻害することでCTADの転写活性を抑制している.すなわち,PHDによってHIFαの発現量が,FIH-1によってHIFαのCTAD転写活性が,共にアミノ酸残基の水酸化によって二重に制御を受けていることになる.ただし,FIH-1の酸素分子に対するKm値は,PHDのそれと比較してかなり低いため6, 10),酸素濃度が低下するにつれ,まずPHDの酵素活性が低下してHIFαのタンパク質発現量が上昇してHIFが活性化し,その後さらに酸素濃度が低下するとFIH-1の酵素活性が低下しCTADにp300が結合し,HIFの転写活性が最大限にまで上昇する.
なお,HIF3αはCTADを欠くこともあり,HIF1αやHIF2αよりも転写活性が低いとされるだけではなく,IPAS, NEPASなど,いくつも知られているスプライシング・バリアント11, 12)のなかにはHIF1αやHIF2αの機能を競合的に阻害するものもある.そのスプライシング制御機構など,HIF3αについてはまだ解明されていない点が多い.
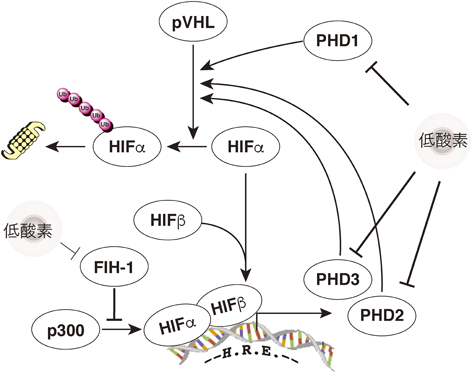
活性化したHIFは,自身を負に制御するはずのPHD3を(細胞種によってはPHD2も)転写する.あたかも,酸素濃度の低下に伴い減弱したPHDの酵素活性を発現量で補おうとしているかのようである.このように,我々の身体にはHIFが恒常的に活性化しないようにするネガティヴ・フィードバック機構が組み込まれていることがわかる(図2).慢性的なHIFの活性化が心筋におけるミトコンドリアの傷害および拡張型心筋症に類似した重篤な心不全を引き起こすこと7, 13, 14),HIFを負に制御する水酸化酵素が三つも遺伝子としてコードされていることからも,どうやら我々の身体は,HIFの恒常的な活性化をなんとかして回避しようとするようにデザインされていることをうかがい知ることができる.
3. PHD依存的代謝制御機構1——全身の細胞における嫌気解糖の活性化——
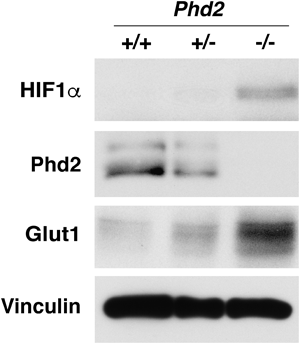
生体内における主要なHIFαのプロリン水酸化酵素であるPHD2の機能が抑えられると,HIFαの発現量が急速に上昇し,HIFが活性化する(図3).HIFはグルコースを細胞に取り込むトランスポーターや多くの解糖系酵素を誘導する.また,ピルビン酸をミトコンドリアTCAサイクルの基質であるアセチルCoAへと変換するピルビン酸デヒドロゲナーゼ(PDH)のE1βサブユニットがHIFによって誘導されるPDHキナーゼ(PDK1など)によってリン酸化されるとPDHの活性が抑制されるため15),HIFの活性化はピルビン酸のTCAサイクルでの利用を妨げる.その結果,細胞に取り込まれたグルコースから代謝されて産生されたピルビン酸は,同じくHIFによって誘導された乳酸デヒドロゲナーゼ(LDH-A)によって乳酸へと変換される.すなわち,HIFの活性化は嫌気解糖を活性化させ,その結果大量の乳酸が細胞外へと放出される2, 16).
4. PHD依存的代謝制御機構2——肝細胞における乳酸クリアランスの活性化——
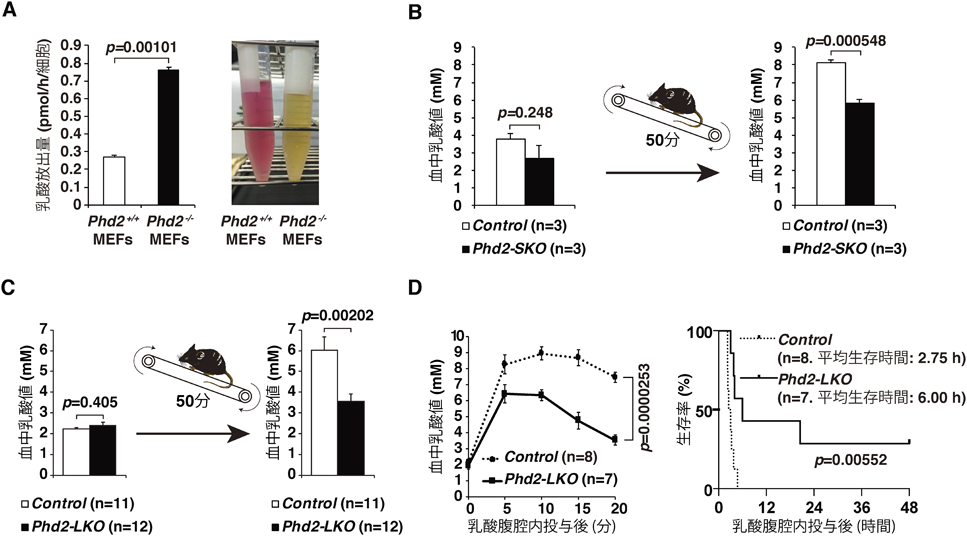
もし全身の細胞において低酸素応答が活性化したら,我々の個体はどうなるであろうか?そこで,低酸素センサー分子PHD2を欠損した細胞やマウスを用いて培養上清や血液中の乳酸を定量した.まず,PHD2を欠損したマウス胎仔性線維芽細胞(MEFs)においては,野生型対照群と比較して乳酸の細胞外への放出速度が有意に速く(図4A,左),培養上清のフェノールレッド試薬が黄色へと変色していることがわかる(図4A,右).この結果から,Phd2遺伝子を全身の細胞で破壊したマウスにおいては,全身の細胞の代謝が嫌気解糖に傾き,尿中に排泄できる量を上回る大量の乳酸が血中に放出され,個体は乳酸アシドーシスに陥ってしまうことが予想された.ところが,タモキシフェン誘導的にPhd2遺伝子を全身の細胞で破壊したマウス(Phd2-SKOマウス)においては,血中乳酸値は予想に反して対照群と比較してむしろ低値であり(図4B,左),また,運動によって生理的に乳酸を負荷すると,驚くべきことに血中乳酸値は対照群よりも有意に低下していた(図4B,右).尿中の乳酸濃度は両群間に差がなかったので,Phd2-SKOマウスでは,腎臓以外のいずれかの臓器が乳酸を処理(代謝)していることを意味している.
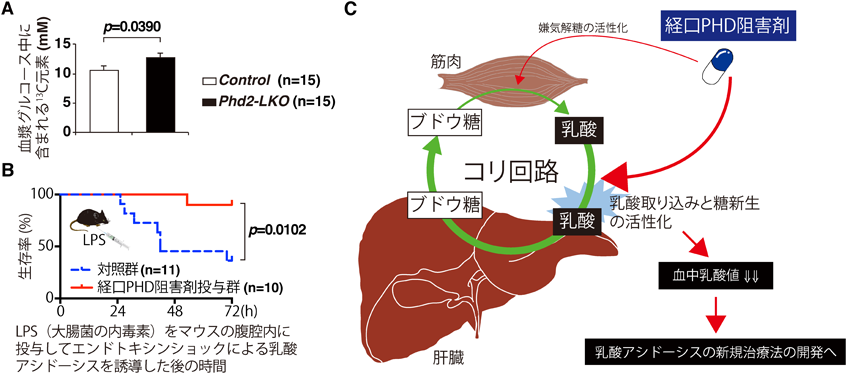
骨格筋細胞などにおける嫌気解糖の結果産生された乳酸は,血流に乗って肝臓に運ばれてグルコースへとリサイクル(糖新生)され,血流に乗って再び骨格筋などへ戻されることが古くから知られている(コリ回路)17)ため,筆者はPhd2-SKOマウスにおいては,乳酸の肝臓への取り込みが亢進しているのではないかと考えた.
そこで,Phd2を肝細胞特異的に破壊したマウス(Phd2-LKOマウス)を作製し,同様に血中乳酸値を測定したところ,Phd2-LKOマウスの安静時の血中乳酸値は対照群と差がなかったが(図4C,左),運動負荷によって生理的に乳酸値を上昇させると,Phd2-LKOマウスの血中乳酸値は対照群と比較して有意に低値であった(図4C,右).すなわち,肝臓でPHD2を抑制すると,乳酸クリアランス能力が強化されることが確認された.
さらに,体外から致死量の乳酸を負荷した乳酸アシドーシスモデルにおいても,Phd2-LKOマウスの血中乳酸値は対照群と比較して低値であり(図4D,左),その後のマウスの生存率も劇的に改善されていた(図4D,右).すなわち,肝臓においてPHD2の機能を抑制すると,致死的乳酸アシドーシスの生存率を改善できることが示された.肝細胞におけるPHD2阻害によって乳酸のクリアランスが改善する理由としては,乳酸を肝細胞内に取り込むモノカルボン酸トランスポーターMCT2や,細胞内に取り込んだ乳酸をピルビン酸へと変換する酵素LDH-Aの発現上昇などが考えられる18).肝細胞に取り込まれた乳酸の代謝経路については,炭素原子すべてを安定同位体13Cで置換した乳酸(13C3乳酸)を対照群およびPhd2-LKOマウスの腹腔内に投与し,その20分後に13C3乳酸から産生された血漿中グルコースを質量分析器で定量したところ,Phd2-LKOマウスの方が対照群と比較して投与した13C3乳酸由来のグルコースが有意に多かったため(図5A),肝細胞においてPHD2の機能を阻害すると,過剰投与した乳酸からの糖新生能力が強化されることが証明された.ただし,これらがすべてPHD2の抑制によるHIFの活性化だけで説明できるものなのかどうか,現時点では不明である
乳酸アシドーシスは,敗血症などのエンドトキシンショックに合併し,生命予後を悪化させる病態である.そこで筆者らは,致死量の大腸菌毒素(LPS:リポポリサッカライド)を投与して作製したエンドトキシンショックモデルマウスを作製し,PHD2の阻害がその生存率を改善させるか否かを調べた.PHD2の阻害剤GSK360Aを経口胃管投与(oral gavage)したマウス群においては,対照群と比較してマウスの生存率の著明な改善が確認できた(図5B).経消化管的に投与された薬剤は,消化管での吸収後門脈を経由してまず肝臓に作用するため,この研究結果は,PHD阻害剤の経口投与が,肝臓における低酸素応答を活性化し,重症感染症などに合併した乳酸アシドーシスにおいて血中乳酸値を低下させて生存率を改善することが可能であることを意味している18).
致死率の高い敗血症などの重篤な感染症の治療成績は血中乳酸値と逆相関するため,重症感染症の治療には原疾患の治療と同時に血中乳酸値を低下させることが必須となる19).今後,肝臓におけるPHD2を介した低酸素応答を標的とした乳酸アシドーシスの新規治療法が,重症感染症などの治療成績を改善することが期待される(図5C).
経口PHD阻害剤はFG-4592(roxadustat)が慢性腎臓病(CKD)患者の腎性貧血治療薬として現在臨床治験中であるが,腎性貧血だけでなく,乳酸アシドーシスへの適応拡大を今後試みたいと思っている.
ただし,先述したとおり,ここで示した肝細胞における乳酸クリアランス機構はPHD依存的なものではあるものの,これが果たしてHIFを介した低酸素応答の結果なのか否かについては現時点では不明である.マスターレギュレーターとも呼ばれるHIFαのクローニング,およびそのプロリン水酸化を介したpVHL依存的ユビキチン–プロテアソーム系によるタンパク質分解機構の解明以来,低酸素応答の多くがPHD–HIF経路で制御されていることが明らかとなったが,PHDにもHIFα以外の基質が同定されつつあり20, 21),PHD依存的だがHIF非依存的な低酸素応答の分子メカニズムについても,今後明らかにされていくものと思われる.
先述したとおり,細胞内にはシトクロムcオキシダーゼやヘムオキシゲナーゼなど,オキシダーゼ・オキシゲナーゼと呼ばれる多数の酸素添加酵素がある.各々の酵素の酸素分子に対するKm値に差こそあれ,これらの酵素は酸素濃度の低下に伴い酵素活性が低下し,さまざまな生理機能がこれらの酵素活性の変動によって制御されており,PHDだけでなくこれらの酵素もみな酸素濃度センサーとして生体の生理機能を制御しているといえよう.たとえば,頸動脈小体における呼吸制御22, 23)や低酸素性肺血管収縮(hypoxic pulmonary vasoconstriction:HPV)24)などは,古くから知られる“低酸素応答”である.JmjCドメインを持つヒストンリシン脱メチル化酵素(KDMs)やメチル化シトシンジオキシゲナーゼ(ten-eleven translocation methylcytosine diokygenase 1–3:TET1–3)などは,PHDやFIH-1と同ファミリーに属する2-オキソグルタル酸・鉄依存的ジオキシゲナーゼであり,低酸素によってその活性が抑制されるため,低酸素応答はエピジェネティクスも制御していることがわかる.また,脳においては,低酸素時にヘムオキシゲナーゼHO-2の活性が低下することで一酸化炭素(CO)の産生量が減少し,COによって活性が抑制されていたシスタチオニンβ合成酵素(CBS)が活性化した結果産生された硫化水素(H2S)によって脳血管が拡張することが明らかとなった25).O2−NO−H2Sという異種ガス状分子のクロストークによって生理機能が複雑に制御されているという事実は大変興味深い.
このように,低酸素応答を正しく掌握するためには,よく研究されているPHDやHIFを介した低酸素応答だけでなく,それ以外の低酸素応答メカニズムをも含めて総合的に理解することが重要である.