クロマチン構造の中心となるヒストンは,アセチル化,メチル化,リン酸化,ユビキチン化などのさまざまな化学的修飾を受け,染色体の高次構造の変化により,ダイナミックに転写を制御し,発生,分化,細胞死やがん化などにおいて重要な役割を果たしていることが明らかとなっている1).これらのヒストン修飾はその組み合わせにより多彩な細胞機能が発揮されることからヒストンコードと呼ばれ,DNA修飾と並んで,重要なエピゲノム構成要素として機能している.ヒストン修飾による転写制御は,化学修飾の種類や修飾を受けるアミノ酸残基により,転写活性化または転写抑制が規定されることが知られている.ヒストンアセチル化は一般に転写活性化に寄与するが2),一方でヒストンメチル化の場合は標的アミノ酸残基の種類と位置により規定されることが多い.たとえば,ヒストンH3の4番目のリシン残基(H3K4)のトリメチル化は転写活性化に働くが,H3K9やH3K27の場合には転写抑制的に寄与するとされている3).他方,本稿で紹介するH3K36のメチル化に関しては,モノ,ジ,トリ(me1, me2, me3)の三つの異なるメチル化状態をとりうるが,これらの修飾には多数のヒストンメチル化酵素,脱メチル化酵素が関与しており,修飾状態が関与する生命現象も多岐にわたっている4).これまで主に表1に示したような,H3K36の脱メチル化を制御するタンパク質とヒト疾患との関連性が報告されており,これらヒストン修飾の生物学的意義を明らかにすることは,その病態の解明に有用な情報を提供すると考えられる.
我々は,Fbxl10 Tgマウスを用いてFbxl10が生体においてがん遺伝子として機能することを明らかにし,その解析過程において,Fbxl10がHSCでミトコンドリアの酸化的リン酸化経路を活性化し,細胞内エネルギー産生を促進させることを見いだした5).本稿では,特にFBXL10と遺伝子発現制御,エネルギー代謝亢進,および発がんとの関連について概説する.その他の個々のヒストンメチル化酵素,脱メチル化酵素の詳細については,優れた総説がすでに発表されているので,そちらをあわせてご参照いただければ幸いである3, 4, 6).
2. ヒストンH3K36修飾とFBXL10の転写制御における役割
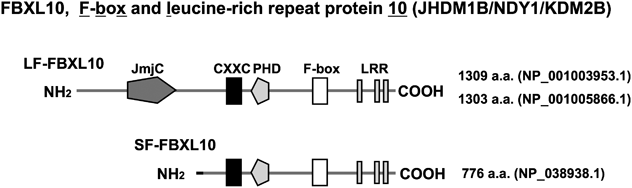
FBXL10(JHDM1B/NDY1/KDM2Bとしても知られる)は約30種類からなるJumonjiファミリーに属するヒストン脱メチル化酵素の一つであり4),N末端よりヒストン脱メチル化酵素活性を持つJmjCドメインに加え,非メチル化CpG DNA結合モチーフであるCXXCドメイン,メチル化ヒストンと相互作用するとされるPHDフィンガードメイン,F-box,タンパク相互作用モチーフであるLRRドメインを有する(図1).FBXL10にはN末端の構造が異なる2種類のスプライシングバリアントが存在し,完全長(long form;LF- FBXL10)とJmjC欠失型(short form;SF- FBXL10)に大別される7–9)(図1).Fbxl10は標的遺伝子に対して,CXXCドメインを介して相互作用することから10),SF-Fbxl10は,LF-FBXL10のDNAへの結合やヒストン脱メチル化酵素活性を競合的に阻害すると想定される.
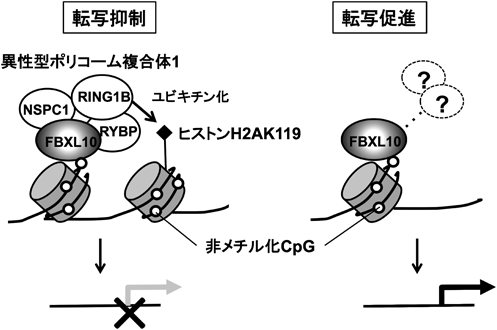
FBXL10は当初,JmjCドメインを持つヒストン脱メチル化酵素として初めて報告されたFBXL11の相同遺伝子として同定され11),その後H3K36me2に特異的な脱メチル化酵素であると報告された12).またH3K4me3に対する脱メチル化活性を有するとも報告されている13).一方,FBXL10には転写抑制因子としての役割が知られており10),最近,転写抑制複合体である異性型ポリコーム複合体1(polycomb repressive complex 1:PRC1)の構成タンパク質の一つとして機能することが明らかにされた14, 15).FBXL11はFBXL10と類似したドメイン構造を有するが,異性型PRC1の構成因子とはなりえず,この役割はFBXL10に特異的である.異性型PRC1は,FBXL10を足場として,非メチル化CpG DNA領域に結合し,ヒストンH2Aの119番目のリシンをユビキチン化(H2AK119Ub)して,転写抑制に機能するとされる15)(図2).異性型PRC1の遺伝子領域への動員とそれに伴うH2AK119Ubには,FBXL10のCXXCまたはLRRドメインが必須であるが,JmjCドメインは必須ではない9, 15).このことは,すべての標的遺伝子座におけるJmjCドメインの機能の関与を否定するものではないが,ヒストン脱メチル化酵素活性は,異性型PRC1の転写抑制能に対して,少なくとも中心的な役割を担ってはいないと推察される.
その一方で,FBXL10は転写活性化へ直接的に関与することが知られている.マウス繊維芽細胞において,FBXL10はJmjCドメイン依存的に抗酸化遺伝子を発現誘導することが報告されている16).また,ヒト膵がん細胞においてヒストンH3K4脱メチル化酵素KDM5Aや転写因子MYCと協調して,翻訳制御因子や細胞内代謝関連遺伝子の活性化に促進的に機能することが知られている17).さらに,iPS細胞へのリプログラミングの際に,ES細胞特異的遺伝子を発現誘導するとも報告されている18).
以上の知見から,FBXL10は状況に応じて,転写抑制にも転写活性にも機能しうると考えられる.最近マウスES細胞におけるFBXL10に対するChIPシークエンスの結果が報告され,1)FBXL10は主に転写開始点近傍(±1 kb)のCpG領域に結合すること,2)FBXL10の結合とH3K36me2レベルの低下は多くの場合共存することが示された14, 15).また,FBXL10の結合様式は転写抑制型,活性型の二つに大別される.すなわち,約5000遺伝子では,Ring1Bといった異性型PRC1の構成因子やH2AK119Ubと共存する一方でRNA Pol IIとは共局在せず転写抑制に関与すると考えられた.逆に,約10,000遺伝子においてFBXL10はRing1BとH2AK119Ubとは独立したゲノム領域に分布しており,H3K4me3やRNA Pol IIと同様に,活性化遺伝子の転写開始点近傍にリクルートされていることが明らかとなった(図2).我々が行ったFbxl10 TgマウスのHSCを用いたChIPシークエンスにおいても,H3K36me2のレベルは,転写開始点±10 kbと比べて転写開始点近傍(±1 kb)で,より低く検出されるという結果を得ている5).これらの報告からFBXL10は,異性型PRC1の構成因子として転写抑制に働く一方で,単独あるいは他のタンパク質と協調して機能する場合には,転写促進的に作用するというモデルが考えられる(図2).
これまでに述べたFBXL10の機能は,発生過程において重要な役割を担っており,その機能欠失は個体発生異常を来すことが知られている.たとえばJmjCドメインを欠失したFbxl10変異マウスでは,一部のマウスが神経管閉鎖不全または外脳症により致死となることが報告されている7).また,DNA結合ドメインであるCXXCドメインを欠失した場合には,Fbxl10変異ホモマウスは胎生期早期致死となり,ヘテロマウスにおいてはホメオティック変異が観察される9)(表1).
表1 主なヒストンH3K36脱メチル化酵素| ヒストン脱メチル化酵素 | 別名 | 報告のある主な基質 | ノックアウトマウス | ヒト疾患との関連 |
|---|
| JMJD5 | KDM8 | H3K36me2 | 胎生致死 | n.d. |
| FBXL10 | JHDM1B, NDY1, KDM2B | H3K36me2, H3K4me3 | 胎生致死,外脳症 | 発現上昇(白血病,膵がん,精巣腫瘍,膀胱がんなど) |
| FBXL11 | JHDM1A, KDM2A | H3K36me1/me2, H3K4me3, p65 NF-κB | 胎生致死 | 発現上昇(肺非小細胞がんなど) |
| JMJD2A | JHDM3A, KDM4A | H3K9me2/me3, H3K36me2/me3, H1.4K26me2/me3 | 心筋負荷ストレス応答の低下 | 発現低下(膀胱がん)発現上昇(乳がんなど) |
| JMJD2B | JHDM3B, KDM4B | 乳腺発育異常 | 発現上昇(末梢神経鞘腫,乳がんなど) |
| JMJD2C | JHDM3C, GASC1, KDM4C | n.d. | 遺伝子重複(食道がん,乳がん)染色体転座(粘膜関連リンパ腫) |
| JMJD2D | JHDM3D, KDM4D | 明らかな異常の報告なし | n.d. |
| NO66 | 2410016O06Rik | H3K4me2/me3, H3K36me2/me3 | n.d. | 発現上昇(肺小細胞がん) |
n.d.: not determined.
これまでに知られているヒストンH3K36脱メチル化酵素の基質と個体レベルでの機能,ヒト疾患との関連(文献3をもとにその後の報告を加えて改変). |
ヒト疾患との関連では,FBXL10の発現上昇とがんとの関連が強く示唆されている.臨床的にはヒト急性骨髄性白血病,リンパ性白血病,精巣腫瘍,膵がん,膀胱がんなどで,FBXL10の高発現が認められる11, 17, 19).
これら発現異常と発がん,がんの進展との関連については実験的にも検証が行われている.Heらは,マウス骨髄移植モデルを用いて,Fbxl10が白血病遺伝子HoxA9/Meis1による白血病の発症・進展を促進し,その機能にはJmjCドメインが必要であることを示した20).また,K-Ras変異を持つマウス膵上皮細胞を用いた免疫不全マウスへの移植モデルにおいても,同様にJmjCドメイン欠失型Fbxl10変異体導入による腫瘍抑制効果が認められている17).これらの解析では,FBXL10はがんの進展に促進的に機能し,これはヒストン脱メチル化酵素活性依存的な機構に起因するとされた.また,ヒストン脱メチル化酵素活性との関連については不明であるが,最近FBXL10は,異性型PRC1の構成因子としてヒト白血病幹細胞の維持に必須であることが報告された.FBXL10を含む異性型PRC1の構成遺伝子をノックダウンしたヒト白血病幹細胞を免疫不全マウスに移植すると,白血病発症に対する顕著な抑制効果が認められた21).
これらの結果はFBXL10が,がん幹細胞の維持,およびがんの進展に促進的に機能することを示している.今回,我々は,HSCsで目的遺伝子を高発現するLy6A/Sca-1プロモーター22)を用いてFbxl10 Tgマウスを作製した.長期観察の結果,Fbxl10 Tgマウスは全例が白血病を発症し,Fbxl10は個体レベルで単独でがん遺伝子として機能することを見いだした.以下にその解析結果について概説する.
4. 骨髄異形成症候群マウスモデルにおけるFbxl10の発現異常の同定とがん遺伝子としてのFbxl10
7番染色体長腕欠損(7q-)症候群は,化学療法後や放射線療法後に発症する白血病や骨髄異形成症候群で高頻度に認められる染色体異常である.我々は以前にマイクロアレイCGH法を用いて同定された疾患責任候補遺伝子Samd9L(SAM domain-containing protein 9-like)の欠損マウスを作製し,長期観察の結果,このマウスが血液細胞の異形成を伴って白血病を発症することを見いだしている23).
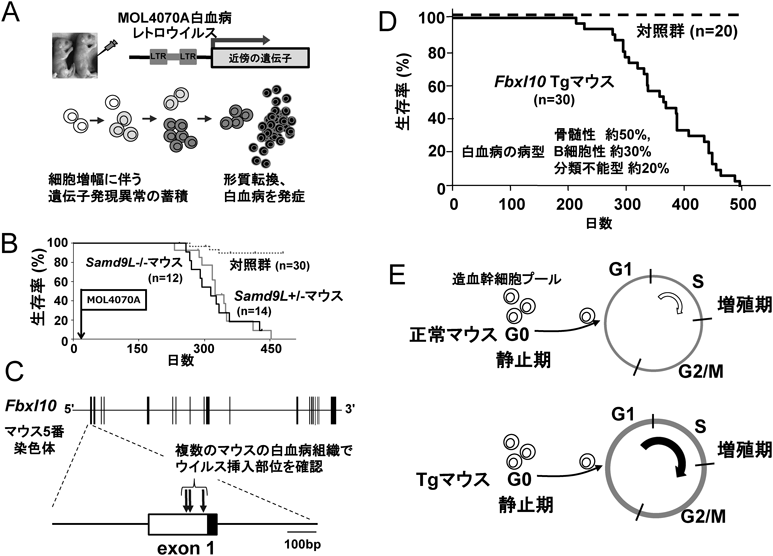
Samd9L欠損マウスの白血病発症は長期の潜伏期を必要とした(約400~500日程度)ことから,二次的な遺伝子異常の関与が強く疑われた.そこでSamd9L欠失と協調して機能する遺伝子を同定する目的で,白血病レトロウイルスを用いた挿入変異解析を行った(図3A).この手法は,新生仔マウス体内に,増幅可能なレトロウイルスを感染させ,レトロウイルスがランダムにマウスゲノムに取り込まれることにより,内在性の遺伝子を活性化(または不活化)することを応用したものである.ウイルス投与を受けたSamd9L欠損マウスは短期間に白血病を発症した(図3B).そこで白血病腫瘍組織(白血化した脾臓)から抽出したDNAを鋳型として,ウイルス特異的なプライマーを用いたPCR法によりウイルス挿入部位を探索したところ,複数のマウスにおいて,ヒストン脱メチル化酵素Fbxl10の上流にウイルスの挿入が検出された(図3C).これらのマウスの白血病腫瘍組織でFbxl10の発現上昇が認められたことから,ウイルスプロモーターにより活性化されたFbxl10遺伝子が,Samd9L欠損と協調的に機能して白血病発症を促進させると考えられた.
Fbxl10の高発現が造血系に与える影響を検討するため,我々はHSCで目的遺伝子を高発現するFbxl10 Tgマウスを作製し,解析を行った5).その結果,Fbxl10 TgマウスはHSCの増殖亢進を伴い,生後7か月から1年半までに骨髄性白血病(約50%),B細胞性リンパ性白血病(約30%)を発症し,全例が致死となることが明らかとなった(図3D).
骨髄においてHSCは多くが静止期にとどまっており,増殖の必要に応じて細胞周期への移行が行われ,最終的には分化へと導かれる.我々はFbxl10 TgマウスによるHSCの細胞周期変化について検討する目的で,白血病の兆候を認める以前の若齢(8週齢)のFbxl10 Tgマウスおよび対照マウスからHSCを単離し,チミジンアナログBrdU(bromodeoxyuridine)の取り込み,および静止期マーカーであるPyronin Y陰性分画の割合について解析を行った.その結果,Fbxl10 TgマウスのHSCはBrdUの取り込みが亢進している一方で,Pyronin Y陰性分画の減少は認められないことが判明した.この結果はHSCにおけるFbxl10の高発現は,HSCプールの枯渇を伴うことなく,細胞周期を亢進させ,HSCの機能亢進に作用することを示している(図3E).さらに骨髄移植を用いた造血再構築能の解析の結果,末梢血においてTgマウス由来の細胞は対照マウス由来の細胞に比較して骨髄球系,およびB細胞系の細胞で高いキメラ率を示した.これはこのTgマウスで観察された白血病の分化系列とも合致する.すなわち,HSCにおけるFbxl10の高発現は,細胞自律的な機構により骨髄球系とB細胞系列への分化優位性ならびに骨髄再構築能の亢進を賦与すると考えられた.
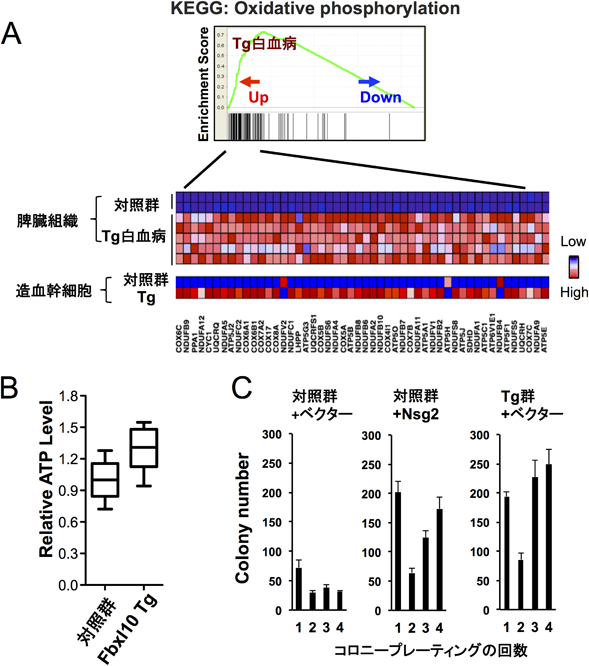
Fbxl10 Tgマウスの表現型における分子基盤を探索するため,若齢のFbxl10 Tgマウスと対照マウスからHSCを単離し,次世代シーケンサーを用いて網羅的な遺伝子発現解析を行った.得られた結果を基に,GSEA(Gene Set Enrichment Analysis)により,KEGG(Kyoto Encyclopedia of Genes and Genomes)パスウェイ上の特定の機能を持つ遺伝子群の発現がどのように変化したかを解析した.その結果,我々はFbxl10 TgマウスのHSCにおいて,ミトコンドリアの酸化的リン酸化経路を構成する遺伝子群の発現が最も上昇していることを見いだした(図4A).同一条件で発現の低下を認めた遺伝子群は検出されなかった.FBXL10抗体を用いて,TgマウスのHSCに対してChIPシークエンスを行ったところ,酸化的リン酸化経路を構成する遺伝子のうち発現上昇が認められたNdufb2(NADH dehydrogenase[ubiquinone]1 beta subcomplex subunit 2, mitochondrial),Ndufs6(NADH dehydrogenase[ubiquinone]iron-sulfur protein 6, mitochondrial),Uqcrfs1(ubiquinol-cytochrome c reductase, Rieske iron-sulfur polypeptide 1)の各遺伝子座において,転写開始点近傍にFBXL10の有意な集積が検出され,これらの三つの遺伝子がFBXL10による直接的標的と考えられた.さらに,白血病を発症したFbxl10 Tgマウスの腫瘍組織を用いて同様の網羅的遺伝子発現解析とGSEAによるパスウェイ解析を行い,若齢マウスと同じく酸化的リン酸化経路が最も発現が上昇している機能遺伝子群であることを見いだした(図4A).これらの結果から,Fbxl10高発現はHSCにおいて酸化的リン酸化経路の亢進を誘導し,この代謝プログラム変化が白血病の発症および腫瘍細胞の維持の両方に重要な働きをしていると考えられた.
増殖が盛んな細胞においては,大量の細胞内エネルギーが必要とされる.細胞内エネルギーであるATPを産生する経路は解糖系,電子伝達系に大別される.解糖系は細胞質で行われる嫌気性代謝であり,電子伝達系はミトコンドリア内膜で行われる好気性代謝である.解糖系では,1分子のグルコースから2分子のATPしか産生されないのに対して,ミトコンドリアの電子伝達系におけるATP産生反応は内膜と外膜の間のプロトン勾配を利用した酸化反応で(酸化的リン酸化),1分子のグルコースから36分子のATPを生み出すことができる.我々は若齢マウスのHSCを用いてそのATP産生について検討を行ったところ,Fbxl10 TgマウスのHSCでは対照群よりも有意に高いATPレベルを認めた(図4B).上記の酸化的リン酸化経路の亢進は,エネルギー代謝を活性化させ,過剰のATPを産生していることが明らかとなった.
がん細胞では,有酸素下であってもミトコンドリアの酸化的リン酸化よりも解糖系を利用してATPを産生するという現象がみられ,ワールブルグ効果(Warburg effect)として知られている24).これは正常細胞と異なるがん細胞の性質として現在まで広く受け入れられている概念である.この現象の意義としては,固形がんはある一定の大きさに成長すると栄養血管新生が追いつかないためその間の低酸素への適応である,あるいは酸化的リン酸化による活性酸素種(reactive oxygen species:ROS)産生に起因する細胞機能障害や遺伝子損傷を防ぐためのものである,など諸説あるが定説には至っていない.
一方で最近の研究から,がん細胞は,解糖系による嫌気的代謝以外にも幅広くエネルギー産生経路を使用することがわかってきた.たとえば,悪性黒色腫において発現上昇が認められるperoxisome proliferator-activated receptor γ coactivator 1-α(PGC-1α)は,酸化的リン酸化を直接的に活性化してエネルギー産生を促す一方で,抗酸化遺伝子の発現誘導にも機能し,ROSの蓄積なくがんの進展に寄与する25).脳神経膠腫で高発現が認められるIGF2 mRNA binding protein 2(Imp2)もまた酸化的リン酸化経路を活性化させてエネルギー産生を亢進させ,腫瘍促進的に作用する.ただし,この場合にはROSの除去能の亢進は認められていない26).酸化的リン酸化によるROS産生は,必ずしもワールブルグ効果の意義を規定するものではない可能性が示唆される.
我々は,酸化的リン酸化経路亢進により蓄積されたROSがDNA損傷を介して白血病発症に関与している可能性を検討する目的で,Fbxl10 TgマウスのHSCに対して,蛍光分子プローブを用いて酸化的リン酸化の活性化によるROS蓄積への影響を検討したが,Fbxl10 TgマウスのHSCsでは対照マウスと比べて有意なミトコンドリアROSの上昇は認められなかった.前述のとおり,FBXL10はマウス繊維芽細胞において抗酸化遺伝子(Aass, Nqo1, Prdx4, Serpinb1b)を誘導し,これらは過酸化水素処理により蓄積する細胞内ROSの除去に機能する16).そこで,これら標的遺伝子を含む種々抗酸化遺伝子群の発現比較をFbxl10 Tgマウスと対照群のHSCとで試みたが,2倍以上の発現変化を示したものは一つも見いだされなかった5, 27).Fbxl10 TgマウスHSCに特有のROS除去機構の存在が推察されるが,現在までその特定には至っていない.
以上の結果から,HSCにおけるFbxl10過剰発現は,ROSの蓄積を伴わず細胞内エネルギー産生を亢進させ,細胞増殖を促し,個体レベルで腫瘍化に促進的に寄与すると考えられた.Fbxl10による酸化的リン酸化経路の活性化は,以前にも膵がん細胞株の解析において報告がある17).我々は,生体においてFbxl10が酸化的リン酸化経路を活性化させ,がん遺伝子として機能することを初めて明らかにした.また最近,FBXL10はヒト白血病幹細胞においても細胞内代謝促進に寄与すると報告されている21).
6. Fbxl10 Tgマウスにおける白血病発症機構
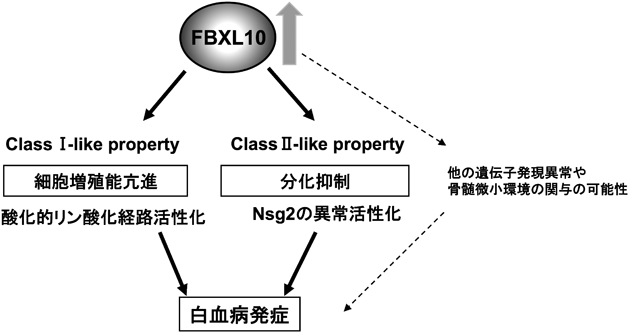
これまで急性白血病に関わる遺伝子変異が数多く報告されているが,Gillilandらは,特に急性骨髄性白血病(acute myelogenous leukemia:AML)の発症には少なくとも2種類の事象が関与するというモデル(急性白血病発症の2段階モデル)を提唱した.すなわち,変異には大別して,細胞の増殖を駆動するclass I遺伝子変異(チロシンキナーゼの機能獲得型変異など)と細胞の分化抑制などに関わるclass II遺伝子変異(分化に関わる転写因子などの変異)という二つのカテゴリーがあり,単独遺伝子の変異では白血病の発症には不十分で,少なくとも両カテゴリーの遺伝子異常を合わせて獲得することが必要とされる28).
我々は,Fbxl10 TgマウスのHSCにおいて個々の遺伝子の発現レベルの変化に着目した.その結果,対照群に比べて約40倍にまで発現上昇を認めた遺伝子として,Nsg2(neuron-specific gene family member 2)を同定した.NSG2はCalyドメインを有する1回膜貫通型タンパク質であり,ドーパミンやグルタミン酸受容体の再利用に機能する神経細胞特異的ファミリー遺伝子の一つである29).通常造血細胞ではその発現が強く抑制されている.Fbxl10 Tgマウスの白血病腫瘍組織でNsg2の発現を調べると特に骨髄性白血病において高い発現が認められた.ChIPアッセイの結果,Fbxl10 TgマウスのHSC,骨髄性白血病細胞ともにNsg2の転写開始点近傍においてFbxl10の高い集積が検出され,これはH3K36me2レベルの低下とも相関していた.Nsg2の発現上昇は,Fbxl10の過剰発現による直接的な影響であると考えられた.
Nsg2を正常対照造血幹細胞・前駆細胞に導入したところ,サイトカイン存在下で分化に抵抗性を示した.また,通常,造血幹細胞・前駆細胞ではリプレーティングにより造血コロニー数は減少するのに対し,Nsg2を導入した細胞では未分化性を反映して,複数回のリプレーティングによっても造血コロニー形成能の維持が確認された(図4C).さらに遺伝子発現データベース検索の結果,FBXL10とNsg2のヒトホモログHMP19はヒト急性骨髄性白血病の約3.4%の症例でともに高発現が認められた.以上のことから,これらの知見はヒト白血病の一部の症例の病態を直接的に反映したものであることが示唆された.
以上,我々の解析から,Fbxl10の造血幹細胞における高発現はエネルギー代謝亢進と分化障害を誘導し,単独でがん遺伝子として白血病発症に関与するものと考えられた(図5).
H3K36メチル化修飾の関与する現象は多岐にわたっているが,その原因としては,この修飾に関与する分子の機能的多様性によるところが大きいと推察される.一つの分子が複数の異なるヒストンメチル化修飾に対して酵素活性を発揮する場合もあれば,他の機能ドメインを介してエピゲノム制御に関与する場合もある.Fbxl10の過剰発現は酸化的リン酸化経路を活性化させ,細胞内エネルギー産生を促進させることが個体レベルで明らかとなったが,その転写制御機構の詳細に関しては不明な点も多い.これらエピジェネティック制御機構の詳細が将来的には明らかにされていくものと考えられる.
謝辞Acknowledgments
本稿の中で述べた著者が関連した実験結果は研究室のメンバーおよび国内外の多くの研究者との共同成果です.すべての共同研究者に感謝いたします.
引用文献References
1) Berdasco, M. & Esteller, M. (2010) Dev. Cell, 19, 698–711.
2) Hayakawa, T. & Nakayama, J. (2011) J. Biomed. Biotechnol., 2011, 129383.
3) Kooistra, S.M. & Helin, K. (2012) Nat. Rev. Mol. Cell Biol., 13, 297–311.
4) Wagner, E.J. & Carpenter, P.B. (2012) Nat. Rev. Mol. Cell Biol., 13, 115–126.
5) Ueda, T., Nagamachi, A., Takubo, K., Yamasaki, N., Matsui, H., Kanai, A., Nakata, Y., Ikeda, K., Konuma, T., Oda, H., Wolff, L., Honda, Z., Wu, X., Helin, K., Iwama, A., Suda, T., Inaba, T., & Honda, H. (2015) Blood, 125, 3437–3446.
6) Klose, R.J., Kallin, E.M., & Zhang, Y. (2006) Nat. Rev. Genet., 7, 715–727.
7) Fukuda, T., Tokunaga, A., Sakamoto, R., & Yoshida, N. (2011) Mol. Cell. Neurosci., 46, 614–624.
8) Inagaki, T., Iwasaki, S., Matsumura, Y., Kawamura, T., Tanaka, T., Abe, Y., Yamasaki, A., Tsurutani, Y., Yoshida, A., Chikaoka, Y., Nakamura, K., Magoori, K., Nakaki, R., Osborne, T.F., Fukami, K., Aburatani, H., Kodama, T., & Sakai, J. (2015) J. Biol. Chem., 290, 4163–4177.
9) Blackledge, N.P., Farcas, A.M., Kondo, T., King, H.W., McGouran, J.F., Hanssen, L.L., Ito, S., Cooper, S., Kondo, K., Koseki, Y., Ishikura, T., Long, H.K., Sheahan, T.W., Brockdorff, N., Kessler, B.M., Koseki, H., & Klose, R.J. (2014) Cell, 157, 1445–1459.
10) Koyama-Nasu, R., David, G., & Tanese, N. (2007) Nat. Cell Biol., 9, 1074–1080.
11) Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M.E., Borchers, C.H., Tempst, P., & Zhang, Y. (2006) Nature, 439, 811–816.
12) He, J., Kallin, E.M., Tsukada, Y.I., & Zhang, Y. (2008) Nat. Struct. Mol. Biol., 15, 1169–1175.
13) Frescas, D., Guardavaccaro, D., Bassermann, F., Koyama-Nasu, R., & Pagano, M. (2007) Nature, 450, 309–317.
14) Farcas, A.M., Blackledge, N.P., Sudbery, I., Long, H.K., McGouran, J.F., Rose, N.R., Lee, S., Sims, D., Cerase, A., Sheahan, T.W., Koseki, H., Brockdorff, N., Ponting, C.P., Kessler, B.M., & Klose, R.J. (2012) eLife, 1, e00205.
15) Wu, X., Johansen, J.V., & Helin, K. (2013) Mol. Cell, 49, 1134–1146.
16) Polytarchou, C., Pfau, R., Hatziapostolou, M., & Tsichlis, P.N. (2008) Mol. Cell. Biol., 28, 7451–7464.
17) Tzatsos, A., Paskaleva, P., Ferrari, F., Deshpande, V., Stoykova, S., Contino, G., Wong, K.K., Lan, F., Trojer, P., Park, P.J., & Bardeesy, N. (2013) J. Clin. Invest., 123, 727–739.
18) Liang, G., He, J., & Zhang, Y. (2012) Nat. Cell Biol., 14, 457–466.
19) Kottakis, F., Polytarchou, C., Foltopoulou, P., Sanidas, I., Kampranis, S.C., & Tsichlis, P.N. (2011) Mol. Cell, 43, 285–298.
20) He, J., Anh, T.N., & Zhang, Y. (2011) Blood, 117, 3869–3880.
21) van den Boom, V., Maat, H., Geugien, M., Rodriguez Lopez, A., Sotoca, A.M., Jaques, J., Brouwers-Vos, A.Z., Fusetti, F., Groen, R.W., Yuan, H., Martens, A.C., Stunnenberg, H.G., Vellenga, E., Martens, J.H., & Schuringa, J.J. (2016) Cell Reports, 14, 332–346.
22) Ma, X.Q., Robin, C., Ottersbach, K., & Dzierzak, E. (2002) Stem Cells, 20, 514–521.
23) Nagamachi, A., Matsui, H., Asou, H., Ozaki, Y., Aki, D., Kanai, A., Takubo, K., Suda, T., Nakamura, T., Wolff, L., Honda, H., & Inaba, T. (2013) Cancer Cell, 24, 305–317.
24) Cairns, R.A., Harris, I.S., & Mak, T.W. (2011) Nat. Rev. Cancer, 11, 85–95.
25) Vazquez, F., Lim, J.H., Chim, H., Bhalla, K., Girnun, G., Pierce, K., Clish, C.B., Granter, S.R., Widlund, H.R., Spiegelman, B.M., & Puigserver, P. (2013) Cancer Cell, 23, 287–301.
26) Janiszewska, M., Suva, M.L., Riggi, N., Houtkooper, R.H., Auwerx, J., Clement-Schatlo, V., Radovanovic, I., Rheinbay, E., Provero, P., & Stamenkovic, I. (2012) Genes Dev., 26, 1926–1944.
27) Gorrini, C., Harris, I.S., & Mak, T.W. (2013) Nat. Rev. Drug Discov., 12, 931–947.
28) Gilliland, D.G. & Tallman, M.S. (2002) Cancer Cell, 1, 417–420.
29) Sabèran-Djoneidi, D., Marey-Semper, I., Picart, R., Studler, J., Tougard, C., Glowinski, J., & Lèvi-Strauss, M. (1995) J. Biol. Chem., 270, 1888–1893.
著者紹介Author Profile
 上田 健(うえだ たけし)
上田 健(うえだ たけし)広島大学原爆放射線医科学研究所疾患モデル解析研究分野助教.博士(医学).
略歴兵庫県神戸市に生る.1996年大阪大学医学部卒業.2005年同大学院医学研究科博士課程修了.06年よりプリンセスマーガレット病院(カナダトロント)博士研究員.10年より現職.
研究テーマ遺伝子改変マウスを用いた固形腫瘍,造血器腫瘍の発症機構の解析.
抱負分子治療標的の探索にも取り組んでみたいと考えております.
ウェブサイトhttp://home.hiroshima-u.ac.jp/sosai/top.html