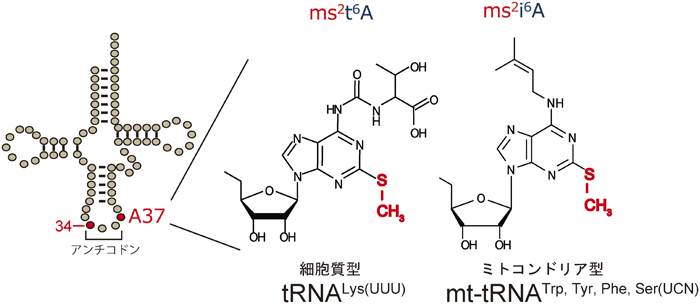
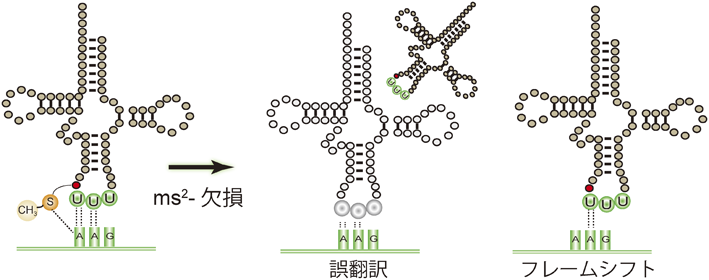
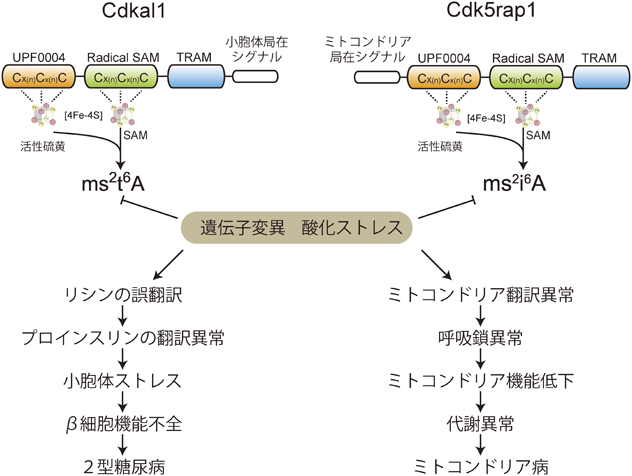
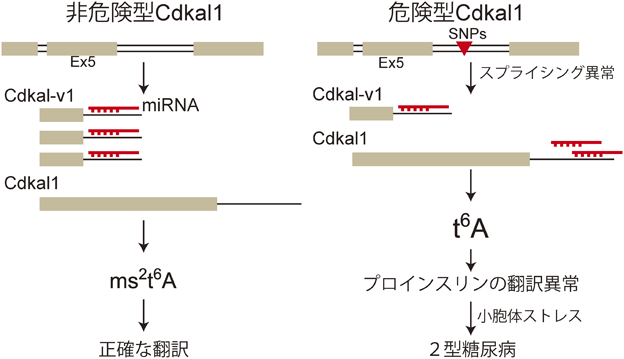
1) Machnicka, M.A., Milanowska, K., Osman Oglou, O., Purta, E., Kurkowska, M., Olchowik, A., Januszewski, W., Kalinowski, S., Dunin-Horkawicz, S., Rother, K.M., Helm, M., Bujnicki, J.M., & Grosjean, H. (2013) Nucleic Acids Res., 41(D1), D262–D267.
3) Arragain, S., Handelman, S.K., Forouhar, F., Wei, F.Y., Tomizawa, K., Hunt, J.F., Douki, T., Fontecave, M., Mulliez, E., & Atta, M. (2010) J. Biol. Chem., 285, 28425–28433.
4) Wei, F.Y., Suzuki, T., Watanabe, S., Kimura, S., Kaitsuka, T., Fujimura, A., Matsui, H., Atta, M., Michiue, H., Fontecave, M., Yamagata, K., Suzuki, T., & Tomizawa, K. (2011) J. Clin. Invest., 121, 3598–3608.
5) Wei, F.Y., Zhou, B., Suzuki, T., Miyata, K., Ujihara, Y., Horiguchi, H., Takahashi, N., Xie, P., Michiue, H., Fujimura, A., Kaitsuka, T., Matsui, H., Koga, Y., Mohri, S., Suzuki, T., Oike, Y., & Tomizawa, K. (2015) Cell Metab., 21, 428–442.
8) Forouhar, F., Arragain, S., Atta, M., Gambarelli, S., Mouesca, J.M., Hussain, M., Xiao, R., Kieffer-Jaquinod, S., Seetharaman, J., Acton, T.B., Montelione, G.T., Mulliez, E., Hunt, J.F., & Fontecave, M. (2013) Nat. Chem. Biol., 9, 333–338.
11) Steinthorsdottir, V., Thorleifsson, G., Reynisdottir, I., Benediktsson, R., Jonsdottir, T., Walters, G.B., Styrkarsdottir, U., Gretarsdottir, S., Emilsson, V., Ghosh, S., Baker, A., Snorradottir, S., Bjarnason, H., Ng, M.C., Hansen, T., Bagger, Y., Wilensky, R.L., Reilly, M.P., Adeyemo, A., Chen, Y., Zhou, J., Gudnason, V., Chen, G., Huang, H., Lashley, K., Doumatey, A., So, W.Y., Ma, R.C., Andersen, G., Borch-Johnsen, K., Jorgensen, T., van Vliet-Ostaptchouk, J.V., Hofker, M.H., Wijmenga, C., Christiansen, C., Rader, D.J., Rotimi, C., Gurney, M., Chan, J.C., Pedersen, O., Sigurdsson, G., Gulcher, J.R., Thorsteinsdottir, U., Kong, A., & Stefansson, K. (2007) Nat. Genet., 39, 770–775.
13) Scott, L.J., Mohlke, K.L., Bonnycastle, L.L., Willer, C.J., Li, Y., Duren, W.L., Erdos, M.R., Stringham, H.M., Chines, P.S., Jackson, A.U., Prokunina-Olsson, L., Ding, C.J., Swift, A.J., Narisu, N., Hu, T., Pruim, R., Xiao, R., Li, X.Y., Conneely, K.N., Riebow, N.L., Sprau, A.G., Tong, M., White, P.P., Hetrick, K.N., Barnhart, M.W., Bark, C.W., Goldstein, J.L., Watkins, L., Xiang, F., Saramies, J., Buchanan, T.A., Watanabe, R.M., Valle, T.T., Kinnunen, L., Abecasis, G.R., Pugh, E.W., Doheny, K.F., Bergman, R.N., Tuomilehto, J., Collins, F.S., & Boehnke, M. (2007) Science, 316, 1341–1345.
14) Zeggini, E., Weedon, M.N., Lindgren, C.M., Frayling, T.M., Elliott, K.S., Lango, H., Timpson, N.J., Perry, J.R., Rayner, N.W., Freathy, R.M., Barrett, J.C., Shields, B., Morris, A.P., Ellard, S., Groves, C.J., Harries, L.W., Marchini, J.L., Owen, K.R., Knight, B., Cardon, L.R., Walker, M., Hitman, G.A., Morris, A.D., Doney, A.S., McCarthy, M.I., & Hattersley, A.T.; Wellcome Trust Case Control Consortium (WTCCC). (2007) Science, 316, 1336–1341.
15) Kirchhoff, K., Machicao, F., Haupt, A., Schäfer, S.A., Tschritter, O., Staiger, H., Stefan, N., Häring, H.U., & Fritsche, A. (2008) Diabetologia, 51, 597–601.
20) Ishikawa, K., Kimura, S., Kobayashi, A., Sato, T., Matsumoto, H., Ujiie, Y., Nakazato, K., Mitsugi, M., & Maruyama, Y. (2005) Circ. J., 69, 617–620.