哺乳類の脳は数千億の神経細胞から構成されているといわれている.神経細胞は,特殊な細胞接着構造であるシナプスを介して互いに接続することで巨大な神経回路を形成し,高次の脳機能を発揮するための情報処理を行う.神経発生過程でのシナプス分化は,シナプスオーガナイザーと呼ばれる膜受容体様接着分子によって誘導される.軸索末端と樹状突起のそれぞれに発現したシナプスオーガナイザーが,細胞外ドメイン(extracellular domain:ECD)を介してトランスシナプティックな選択的相互作用をすることにより,前シナプスと後シナプスの形成を誘導する.シナプスオーガナイザーは,シナプス標的選択の鍵を握るとともに,興奮性シナプスと抑制性シナプスのバランスを調節する役割を担うことも示唆されている.このバランス調節の破綻は自閉症や知的障害などの神経発達障害に関係していると考えられており,実際に,シナプスオーガナイザーをコードする遺伝子の異常が神経発達障害と関連することが報告されている1–6).
前シナプスのオーガナイザーとしてNeurexin(Nrxn)とIIa型受容体チロシンホスファターゼ(receptor protein tyrosine phosphatase:RPTP)の2種類のタンパク質ファミリーが知られている3, 5, 6).Nrxnにはαとβの2種類のアイソフォーム(α-Nrxnおよびβ-Nrxn)が存在する.α-NrxnのECDは六つのlaminin/Nrxn/sex-hormone-binding globular(LNS)ドメインと三つの上皮成長因子(epidermal growth factor:EGF)様のドメインを持つが,二つのLNSドメインにはさまれたEGF様ドメイン(LNS-EGF-LNS)を一つの構造単位として,3単位の繰り返しとして構成される.一方,β-NrxnのECDは一つのLNSのみで構成される.β-NrxnのLNSは,α-Nrxnの6番目のLNSに対応する.Nrxnと相互作用する後シナプスのオーガナイザーとしては,イオンチャネル型グルタミン酸受容体ファミリーのGluD27),ロイシンリッチリピート(leucine-rich repeat:LRR)膜貫通タンパク質(LRR transmembrane proteins:LRRTM)8)とNeuroligin(Nlgn)3)の各タンパク質ファミリーが知られている.これらのうちGluD2は,分泌タンパク質Cbln1を介してNrxnと相互作用する.
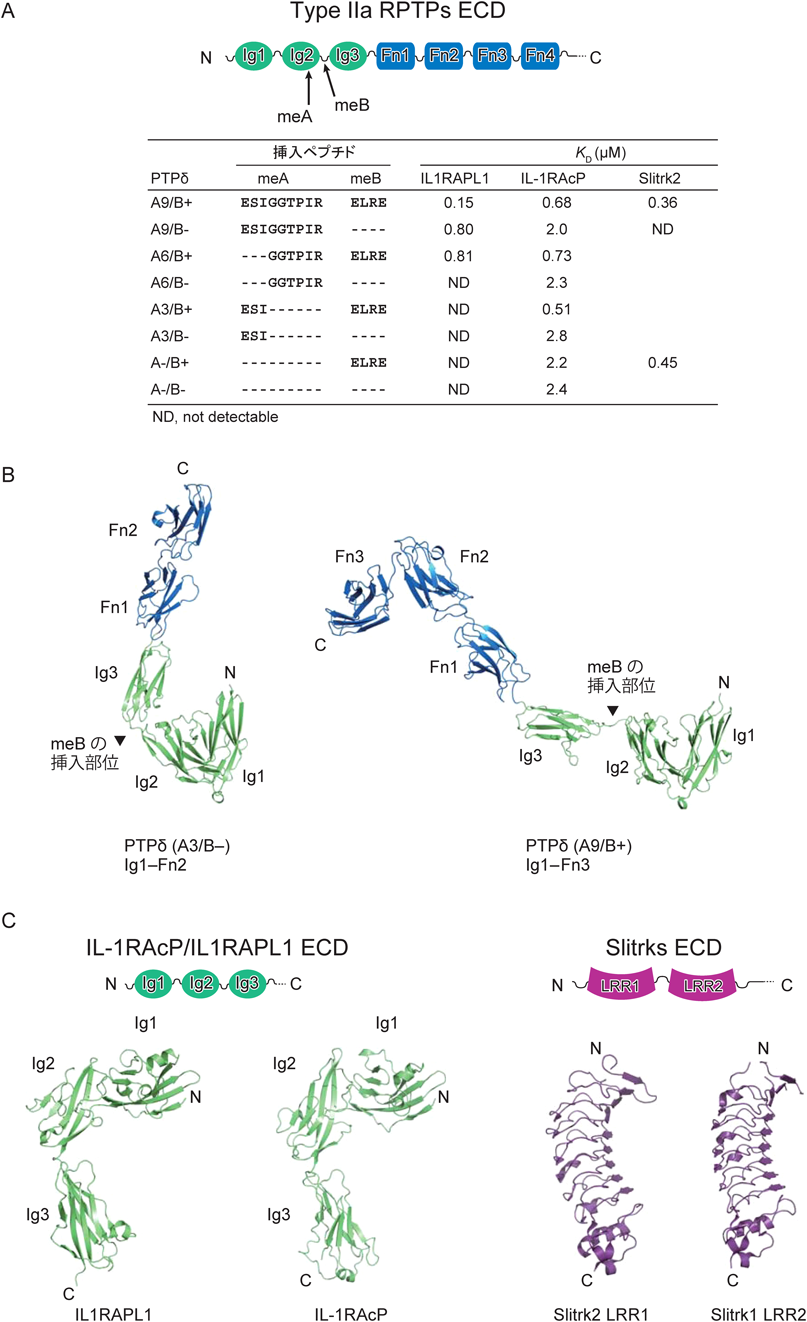
IIa型RPTPは,哺乳類ではPTPδ, PTPσ, LARの3種類がある5, 6).IIa型RPTPのECDは,三つの免疫グロブリン(Ig)ドメイン(Ig1~3)と四つから九つのIII型フィブロネクチン(Fn)ドメイン(Fn1~9)がN末端側から順に並んだ形で構成される(図1A).IIa型RPTPは,後シナプスのオーガナイザーであるインターロイキン1受容体アクセサリータンパク質(interleukin-1 receptor accessory protein:IL-1RAcP)9),IL-1RAcP様1(IL-1RAcP-like1:IL1RAPL1)10),TrkC11),ネトリンGリガンド-3(Netrin G ligand 3:NGL-3)12)や,SlitおよびTrk様(Slit- and Trk-like:Slitrk)タンパク質ファミリー13)と相互作用してシナプス形成を誘導する.
2. 選択的スプライシングによるシナプス標的の調節
後シナプスのオーガナイザーの多くは,選択的スプライシングによって生じるNrxnやIIa型RPTPのバリアントのそれぞれに対して異なる結合能を示す.たとえば,後シナプスのオーガナイザーとの結合能を制御するNrxnのスプライス部位は,α-Nrxnとβ-Nrxnに共通のLNSドメインに存在するスプライス部位4(SS-4)であり,Cbln1–GluD2はSS-4に挿入のあるバリアントと選択的に結合するのに対して,LRRTMはSS-4に挿入のないバリアントと選択的に結合する7, 8).一方,後シナプスのオーガナイザーとの結合能を制御するIIa型RPTPのスプライス部位は,Ig2内およびIg2とIg3の境界に存在する(図1A).この2か所に短いエクソン(ミニエクソン)に対応するペプチドが挿入される.前者はミニエクソンペプチドA(mini-exon peptide A:meA),後者はミニエクソンペプチドB(mini-exon peptide B:meB)と呼ばれる.PTPδの場合は,9残基,6残基,3残基のmeAが挿入されたバリアント(A9, A6, A3)と欠失したバリアント(A−)が存在する.さらに各々のmeAのバリアントに,4残基のmeBが挿入されたバリアント(B+)と欠失したバリアント(B−)が存在する.たとえば,同じIL-1RファミリーのIL1RAPL1とIL-1RAcPであっても,結合特異性はバリアントによって異なる(図1A)9, 10, 14).IL1RAPL1はA9とB+のバリアント(A9/B+)に最も強く結合し,A6/B+やA9/B−にもやや弱く結合するが,その他のバリアントとは結合しない10, 14).一方,IL-1RAcPはA9/B+, A6/B+, A3/B+に対してほぼ同じ強さで結合するが,meAやmeBの欠失によって結合が弱くなる9, 14).また,Slitrkは,meAの有無や配列とは無関係に,B+だけに選択的に結合する15, 16).
最近,我々の報告も含めて,IIa型RPTPのシナプスオーガナイザー複合体の結晶構造が複数報告され,IIa型RPTPの選択的スプライシングによるシナプス標的の調節メカニズムの構造基盤が明らかになったので本稿で紹介する14–16).
3. IIa型RPTPのシナプスオーガナイザー複合体の立体構造
最初に,IIa型RPTPのPTPδとその相互作用相手であるIL1RAPL1, IL-1RAcP, Slitrk1およびSlitrk2それぞれのECDの概観にふれておきたい.図1Bに示すように,PTPδのECDは細長く伸びた形をしている14).N末端側に位置するIg1とIg2は,ドメイン間の相互作用により,コンパクトなV字形の構造ユニットを形成する.それに続くIg3は,meBが挿入されない場合はIg2の近傍に位置することになるが,meBが挿入されるとIg1–2のユニットからは空間的に離れて位置することができる.Ig3, Fn1, Fn2は直線状に並ぶが,Fn3の前で折れ曲がる.IL1RAPL1とIL-1RAcPのECDは三つのIgドメインで構成され,L字形に配置されている(図1C)14).PTPδとの複合体では,先端に位置するIg1がPTPδのIg2およびIg3の両方と相互作用する.さらに,IL1RAPL1では,Ig3がPTPδのIg1と相互作用する.SlitrkのECDは,二つのLRRドメイン(N末端から順にLRR1, LRR2)で構成される(図1C).LRR1とLRR2ともにリピート数が比較的少ないLRRであり,扇形をしている15, 16).LRR1がIIa型RPTPとの結合を担っており,前シナプスの形成を誘導することができる15, 16).
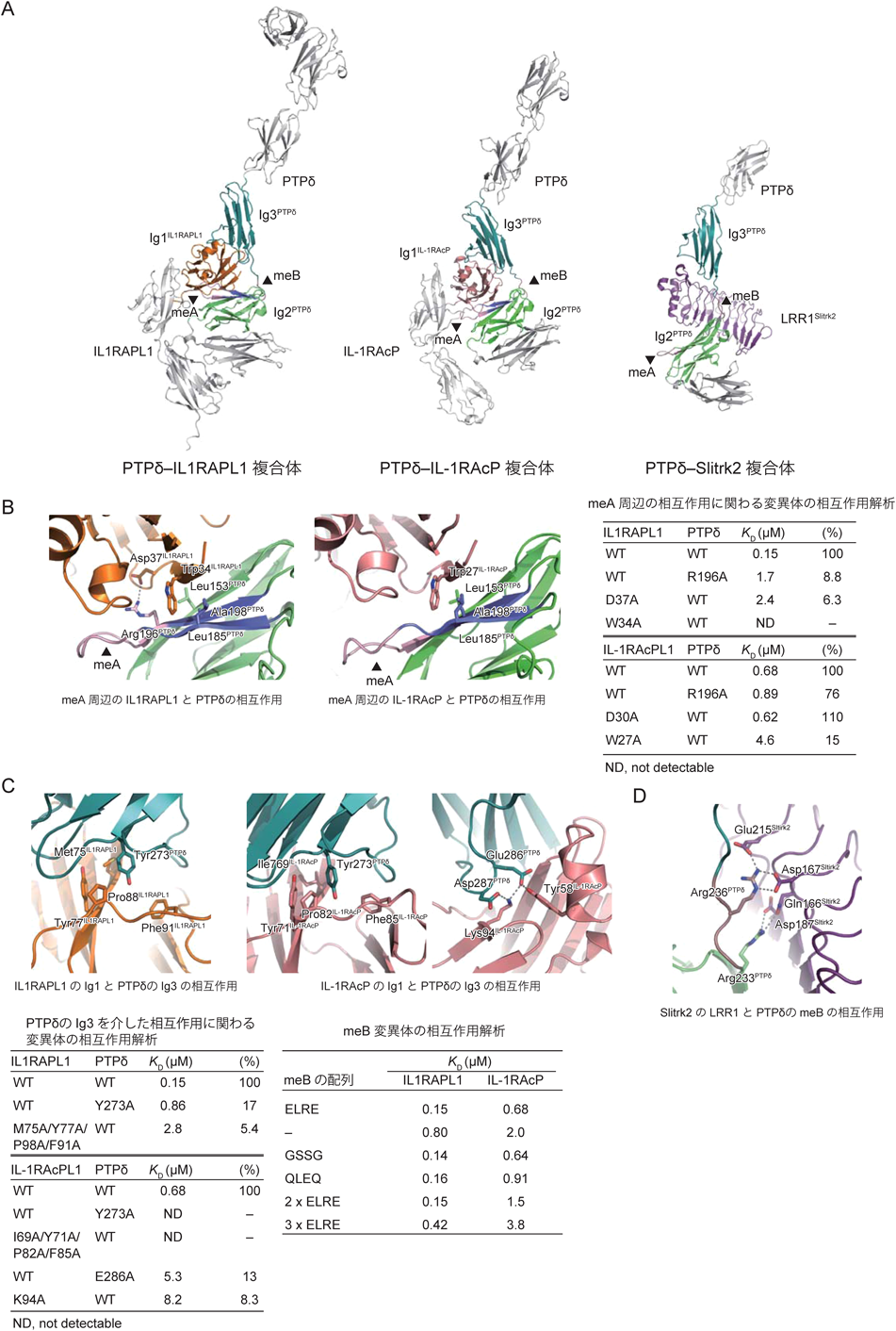
PTPδのIg2に挿入されているmeAは,IL1RAPL1やIL-1RAcPのIg1と相互作用する2本のβストランドをつなぐターンを形成する(図2A, B).この相互作用部位ではIL1RAPL1のTrp34やIL-1RAcPのTrp27がPTPδのLeu153, Ala198やLeu185と疎水性の相互作用をしている(図2B).それに加えて,IL1RAPL1との結合では,A9やA6のC末端のアミノ酸残基であるArg196とIL1RAPL1のAsp37の間に水素結合が形成されている.Arg196の側鎖の疎水部は,IL1RAPL1のTrp34と相互作用することで疎水性の相互作用を強めている.一方,IL-1RAcPとの複合体では,IL1RAPL1のAsp37に相当するAsp30の近くにPTPδのArg196が位置しているものの,側鎖の電子密度がみえておらず,水素結合は形成されていない.結果的に,IL1RAPL1との複合体でみられたようなArg196による疎水性相互作用の強化は起きていない.
これらの構造的知見は,部位特異的変異体を用いた相互作用解析の結果と一致する(図2B).IL1RAPL1のW34A変異体は結合を失うのに対して,IL-1RAcPのW27A変異体は親和性が1/7に低下するが結合能は残る.また,PTPδのR196A変異はIL1RAPL1との結合を著しく弱めるが,IL-1RAcPとの結合にはほとんど影響しない.同様に,IL1RAPL1のD37A変異体も結合を著しく減じるのに対して,IL-1RAcPのD30A変異体は野生型同様の結合能を持つ.PTPδのIg2を介した相互作用は,IL1RAPL1との結合では必須であり,PTPδのArg196とIL1RAPL1のAsp37間の水素結合がその調節の鍵を握っている.そのため,Arg196を含むバリアントであるA9もしくはA6のみが結合能を有する.一方,IL-1RAcPとの結合では,PTPδのArg196はその調節には関わらない.したがって,A9, A6, A3のどのバリアントでも同様の結合能を有する.
PTPδがIL1RAPL1やIL-1RAcPと結合する際に,meBはIL1RAPL1やIL-1RAcPと直接相互作用して認識されているわけではない(図2A).最初にPTPδの構造の説明の際にもふれたが,B+とB−のバリアントの構造を比較してみると,meBの有無でIg1–2のユニットに対するIg3の配置に違いが生じることがわかる(図1B).PTPδのIg3はIL1RAPL1やIL-1RAcPのIg1と相互作用する(図2A, C).この相互作用は,PTPδのTyr273とIL1RAPL1のTyr77あるいはIL-1RAcPのTyr71をコアとした疎水性相互作用である.IL-1RAcPとの結合の場合は,これらのチロシン残基が主鎖との水素結合によって強化されている.さらに,PTPδのIg3のGlu286とAsp287がIL-1RAcPのLys94とTyr58のそれぞれと水素結合を形成している.PTPδのIg3とIL-1RAcPのIg1との疎水性相互作用に影響するPTPδのY273A変異体やIL-1RAcPの疎水性残基群の多重変異体(I69A/Y71A/P82A/F85A変異体)が完全に結合を失うのに対し,IL1RAPL1のIg1との疎水性相互作用に影響する同様の変異体(M75A/Y77A/P98A/F91A変異体)は弱い結合を維持する(図2C).一方,PTPδのIg3とIL-1RAcPのIg1との水素結合に影響するIL-1RAcPのK94A変異体やPTPδのE286A変異体では結合が10倍近く落ちるが,完全には結合が失われないことから,PTPδとIL-1RAcPの結合を強める役割を果たしていると考えられる.構造的な知見とこれらの変異体解析の結果は,PTPδのIg3との相互作用がIL-1RAcPとの結合に必須であるのに対して,IL1RAPL1との結合には重要ではあるが,必須ではないことを示している.
以上の結果から,meBは,PTPδのIg2とIg3が同時にIL-1RAcPやIL1RAPL1のIg1と相互作用できるように適切に配置させるためのリンカーとして機能することが考えられた.実際に,meBの4残基の配列をELREからGSSGやQLEQへと変化させても結合に影響はないが,meBと同じ配列を3回繰り返して12残基に伸ばした場合には結合能が低下した.これらの結果はmeBが結合に適した長さのリンカーであることを支持している.
一方,Slitrkとの結合においては,meBは直接の相互作用に関与する(図2D).Slitrk1および2との複合体の構造では,LRR1がmeBのArg236と水素結合を形成することによりPTPδのmeBを直接認識している15, 16).Slitrk2ではAsp167とGlu215が水素結合を形成している.Arg236との水素結合は必須であり,PTPδのR236E変異体,Slitrk2のD167A変異体は結合を失い,E215A変異体は解離定数を決定できないレベルに結合能が低下する.
シナプスオーガナイザーによるシナプス形成の誘導は,シナプスオーガナイザーを発現する非神経細胞やシナプスオーガナイザーをコートしたビーズを初代培養神経細胞と共培養し,前シナプスや後シナプスに特異的な分子マーカーの集積を調べることで確認することができる.このアッセイ法を利用して,本稿で紹介したPTPδとIL-1RAcP, IL1RAPL1, Slitrk1および2との複合体構造から明らかになった相互作用が,複合体形成とそれに続くシナプス誘導に重要であることも示されている14–16).結合の強さとシナプス誘導能には正の相関がみられるが,シナプス誘導に必要な結合力にはある一定の閾値が存在することが示唆された.これは,非特異的な結合による誤ったシナプス誘導を防ぐ意味合いがあるものと思われる.シナプスオーガナイザー複合体の形成とそのシナプス誘導能の関係が具体的に理解できた一方で,複合体形成に続くイベントがどのようなものであるかはほとんどわかっていない.下流シグナルも含めた分子機構の理解が今後の課題である.
引用文献References
1) Carrié, A., Jun, L., Bienvenu, T., Vinet, M.C., McDonell, N., Couvert, P., Zemni, R., Cardona, A., Van Buggenhout, G., Frints, S., Hamel, B., Moraine, C., Ropers, H.H., Strom, T., Howell, G.R., Whittaker, A., Ross, M.T., Kahn, A., Fryns, J.P., Beldjord, C., Marynen, P., & Chelly, J. (1999) Nat. Genet., 23, 25–31.
2) Jamain, S., Quach, H., Betancur, C., Råstam, M., Colineaux, C., Gillberg, I.C., Soderstrom, H., Giros, B., Leboyer, M., Gillberg, C., & Bourgeron, T. (2008) Nat. Genet., 34, 27–29.
3) Sudhof, T.C. (2008) Nature, 455, 903–911.
4) Pinto, D., et al. (2010) Nature, 466, 368–372.
5) Takahashi, H. & Craig, A.M. (2013) Trends Neurosci., 36, 522–5334.
6) Um, J.W. & Ko, J. (2013) Trends Cell Biol., 23, 465–475.
7) Uemura, T., Lee, S.J., Yasumura, M., Takeuchi, T., Yoshida, T., Ra, M., Taguchi, R., Sakimura, K., & Mishina, M. (2010) Cell, 141, 1068–1079.
8) Siddiqui, T.J., Tari, P.K., Connor, S.A., Zhang, P., Dobie, F.A., She, K., Kawabe, H., Wang, Y.T., Brose, N., & Craig, A.M. (2010) J. Neurosci., 30, 7495–7506.
9) Yoshida, T., Shiroshima, T., Lee, S.J., Yasumura, M., Uemura, T., Chen, X., Iwakura, Y., & Mishina, M. (2012) J. Neurosci., 32, 2588–2600.
10) Yoshida, T., Yasumura, M., Uemura, T., Lee, S.J., Ra, M., Taguchi, R., Iwakura, Y., & Mishina, M. (2011) J. Neurosci., 31, 13485–13499.
11) Takahashi, H., Arstikaitis, P., Prasad, T., Bartlett, T.E., Wang, Y.T., Murphy, T.H., & Craig, A.M. (2011) Neuron, 69, 287–303.
12) Woo, J., Kwon, S.K., Choi, S., Kim, S., Lee, J.R., Dunah, A.W., Sheng, M., & Kim, E. (2009) Nat. Neurosci., 12, 428–437.
13) Yim, Y.S., Kwon, Y., Nam, J., Yoon, H.I., Lee, K., Kim, D.G., Kim, E., Kim, C.H., & Ko, J. (2013) Proc. Natl. Acad. Sci. USA, 110, 4057–4062.
14) Yamagata, A., Yoshida, T., Sato, Y., Goto-Ito, S., Uemura, T., Maeda, A., Shiroshima, T., Iwasawa-Okamoto, S., Mori, H., Mishina, M., & Fukai, S. (2015) Nat. Commun., 6, 6926.
15) Um, J.W., Kim, K.H., Park, B.S., Choi, Y., Kim, D., Kim, C.Y., Kim, S.J., Kim, M., Ko, J.S., Lee, S.G., Choii, G., Nam, J., Heo, W.D., Kim, E., Lee, J.O., Ko, J., & Kim, H.M. (2014) Nat. Commun., 5, 5423.
16) Yamagata, A., Sato, Y., Goto-Ito, S., Uemura, T., Maeda, A., Shiroshima, T., Yoshida, T., & Fukai, S. (2015) Sci. Rep., 5, 9686.
著者紹介Author Profile
 深井 周也(ふかい しゅうや)
深井 周也(ふかい しゅうや)東京大学准教授(放射光連携研究機構/分子細胞生物学研究所).博士(理学).
略歴1974年広島県に生る.97年東京大学理学部生物化学科卒業.99年同大学院理学系研究科生物化学専攻修士課程修了.同年日本学術振興会特別研究員DC1. 2002年東京大学大学院理学系研究科生物化学専攻博士課程修了.同年日本学術振興会海外特別研究員.03年東京工業大学大学院生命理工学研究科助手.06年同大学バイオ研究基盤支援総合センター助教授.07年より現職.
研究テーマと抱負構造神経科学,ユビキチンシグナルの構造生物学.
ウェブサイトhttp://www.iam.u-tokyo.ac.jp/srro/
趣味壁登り.
 山形 敦史(やまがた あつし)
山形 敦史(やまがた あつし)東京大学助教(放射光連携研究機構/分子細胞生物学研究所).博士(理学).
略歴1976年三重県に生る.98年大阪大学理学部生物科学科卒業.2000年同大学院理学研究科生物科学専攻修士課程修了.01年日本学術振興会特別研究員DC2. 02年大阪大学大学院理学研究科生物科学専攻博士課程修了.同年米国スクリップス研究所博士研究員.07年より現職.
研究テーマと抱負シナプス形成の構造生物学.
ウェブサイトhttp://www.iam.u-tokyo.ac.jp/srro/
趣味山登り,キャンプ.