2. タンパク質構造生物学研究へのSAIL法の展開
高分子量タンパク質においては,立体構造決定に必要なNMR構造情報の取得が困難となることは先に述べた.SAIL法はこれら問題を克服するための優れた手法ではあるが,結晶構造既知のタンパク質を対象とする場合には,NMR法により立体構造を再度決定する必要は必ずしもない.むしろ,タンパク質複合体や膜タンパク質においては,タンパク質中の特定のアミノ酸残基のNMRシグナルをプローブとして,他の手法では得にくい構造動態情報を入手することの方がより重要であろう.このため,高分子量タンパク質の特定部位に関するNMRシグナルの高感度測定・帰属技術開発には多くの興味が集中し,1H–15N TROSY(transverse relaxation optimized spectroscopy)法はその先駆けとなった2).本手法では,高分子量タンパク質中の窒素を均一に15N標識し,また炭素原子に結合する水素を均一に重水素置換したタンパク質試料が利用される.このような同位体標識高分子量タンパク質の1H–15N相関スペクトルには,異なった緩和機構が相殺するために1H, 15N軸の線幅が著しく狭い成分が含まれる.TROSYシグナルと呼ばれるこのような成分を選択的に検出することにより,100 kDaを超えるような高分子量のタンパク質の主鎖1H–15Nシグナルが高感度・高分解能で観測できるようになったことは大きな進歩であった2).1H–15N TROSY法に続いて,Ile/Leu/Val(以下,ILVと略称)残基のメチル基のみを13CH3にし,それ以外をすべて重水素化したタンパク質を用いることにより,鋭い1H–13C HMQCシグナルを観測する手法が“Methyl TROSY”法として報告され,高分子量タンパク質へのNMR法の応用範囲が一挙に拡大した3, 4).このようにアミノ酸残基の主鎖アミド基やメチル基のNMRシグナルに限定されてはいても,それらのアミノ酸配列上の部位が帰属できれば,タンパク質の立体構造や動態に関する情報をもたらすプローブとして有用である.このような“NMRの窓”をさらに拡げることにより,立体構造決定自体は困難であるような高分子量タンパク質複合体についても,それらの局所的構造の揺らぎ,あるいはタンパク質間やタンパク質-リガンド間の相互作用動態に関する貴重な情報が得られる可能性がある.NMR法に期待される構造生物学への寄与が,溶液内における相互作用・動態解析に傾きつつあるなかで,メチル基,主鎖アミド基以外のさまざまなNMRの窓を高分子量タンパク質に開くことは最先端の安定同位体利用NMR技術としてのSAIL法に託された重要な開発課題である.このような背景から,我々は高分子量タンパク質に含まれる芳香環や脂肪族側鎖を含めた,すべてのNMRシグナルを選択的,かつ高感度に観測・帰属するためにSAIL法のさらなる改良に取り組んできた.
1)高分子量タンパク質中のLeu/Valメチルシグナルの立体選択的観測
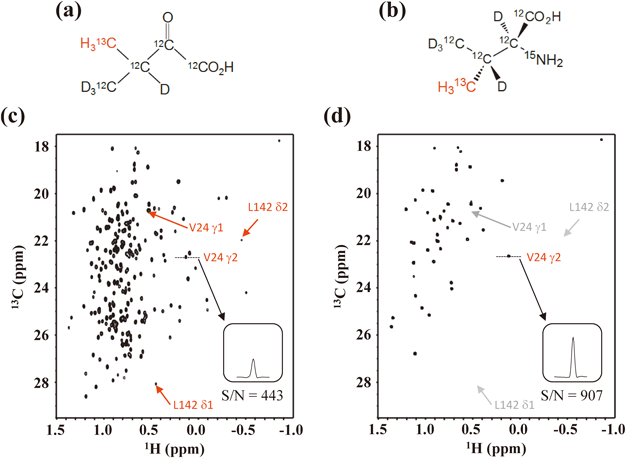
タンパク質には,メチル基を持つアミノ酸残基(Ile, Leu, Val, Ala, Thr, Met)が数多く含まれているため,これらの残基のメチル基(13C1H3)シグナルは,タンパク質全体の立体構造変化や分子間相互作用を俯瞰するための格好のNMRの窓を提供する.現在では,ILV残基のみではなく,Ala, Thr, Metを含めたすべてのメチル基のNMRシグナルがNMRの窓として利用されている5–11).ILV残基はタンパク質の疎水性領域を構成する主要なアミノ酸であり,またそれらのメチル基は他のシグナルとは比較的離れた化学シフトを持ち観測上有利なために有用性は高い.ILVメチル基を残基選択的,および位置・立体選択的に同位体標識する技術開発に多くの関心が寄せられているのはこの理由による.タンパク質中の特定のアミノ酸残基のみを選択的に同位体標識するためには,標識アミノ酸を発現用培地に添加する方法が伝統的に用いられてきた.しかしながら,ILVに各2か所含まれるメチル基を位置・立体選択的に13C標識したILVの入手が困難であったために,合成が容易な同位体標識生合成中間体を用いる方法が広く利用されている.たとえば,α-ケトイソ吉草酸(α-ketoisovalerate:α-KIV)がVal/Leuのメチル基の13C標識前駆体として,α-ケト酪酸(α -ketobutyric acid:α-KB)がIleのδ1メチル基の13C標識前駆体として開発され,市販もされている5–8).ValとLeuのプロキラルメチル基の一方を12CD3,他方を13CH3とした[4-13CH3]-α-KIV(図1a)を重水素化培地中に添加すれば,Valを経由して生合成されるLeuも同時に標識されるために,タンパク質中のすべてのVal/Leu残基のγ1/γ2-,およびδ1/δ2-メチル基が13CH3標識された重水素化タンパク質試料が得られる.このように,立体非特異的にプロキラルメチル基の一方のみを標識した[4-13CH3]-α-KIVを前駆体として調製したリンゴ酸合成酵素(malate synthase G:MSG)の1H-13C HMQCスペクトルを図1cに示す.MSGは723残基からなる82 kDaの高分子量タンパク質であり,LeuとValをそれぞれ70および46残基を持つために,計算上は232個の1H-13C相関シグナルが観測されるはずである.実際に得られた1H-13C HMQC(hetero-nuclear multiple quantum coherence)スペクトルにおける1H–13C相関シグナルの分離はきわめて良好である.
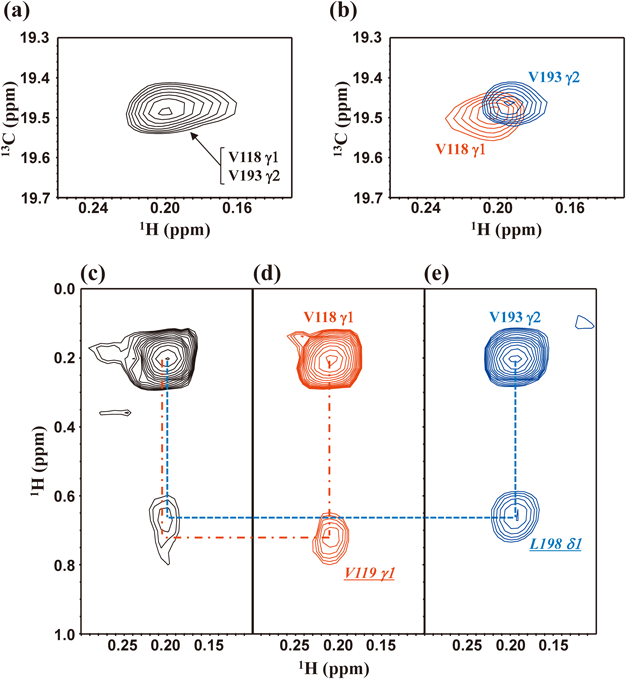
このように,メチル基を立体非特異的に同位体標識したα-KIVをLeu/Valの標識前駆体として用いる方法は経済性,実用性に優れた方法であるが,欠点もある.すなわち,Val/Leu残基のプロキラルメチル基は非選択的に一方が13CH3,他方が12CD3となるために,それらのシグナルの立体選択的帰属が別途必要となり,またVal/Leuのγ1/γ2-,およびδ1/δ2-メチル基の標識率が50%以下にとどまる点も問題である.このような問題点は,同位体標識中間体の代わりに,立体選択的にメチル基を同位体標識したValおよびLeuを用いることにより完全に解決できる12).たとえば,[4-13CH3]-α-KIVの代わりに,γ2-メチル基のみを立体選択的に標識した“γ2-Val”(図1b)を用い,さらにValからLeuへの生合成経路を抑制するために,十分な量の重水素化Leu, [U-D]-Leuを添加することにより,Valのγ2-メチル基のみを立体選択的標識したMSGを調製することができる12).このようにして得られた標識MSGのNMRスペクトルにおいては,メチル基領域に46残基のValに由来するγ2-メチルシグナル以外,Valのγ1-メチルシグナル,Leuのδ1/δ2-メチルシグナルはまったく観測されない(図1d).同一条件下で測定したメチルシグナルの強度をVal24のγ2について比較したところ,γ2-Valを用いた標識MSGでは予想どおり約2倍の感度向上が得られた.同様に,プロキラルメチル基の一方のみを13CH3標識した“γ1-Val”,“δ1-Leu”,“δ2-Leu”を単独,あるいは組み合わせて標識することにより,標識中間体を用いては不可能な組み合わせで残基・立体特異的に高濃度標識したタンパク質試料の調製が可能となる.立体選択的に標識アミノ酸を用いれば,プロキラルメチル基の立体帰属の不確定さは避けられ,倍以上の同位体標識率が得られるために,立体構造決定に必要なNOE情報を迅速,かつ高精度に得ることができる(図2).α-KIVを用いて標識したMSGでは,図2aの1H13C相関シグナルはValのメチル基に帰属されるが,その線形から単一のシグナルによるものではないと推定される.事実,図2bに示すように,“γ1-Val+δ2-Leu”,および“γ2-Val+δ1-Leu”の組み合わせで標識したMSGの当該部分のシグナル(それぞれ,赤と青で表示)を比較すると,α-KIV標識で得られるシグナルは,ほぼ重なり合ったV118 γ1とV193 γ2-メチルシグナルに由来することが判明した.このように重なり合ったシグナルを個別に観測できることは,立体構造情報としてのNOEの価値を高める.すなわち,標識α-KIV中間体を用いて調製した非立体選択的メチル標識MSGでは,重なり合ったV118 γ1とV193 γ2-メチルシグナルに近接したメチル基を特定するためのΝΟΕシグナルは図2cのように重なり合うために解析不能であるが,γ1-Val+δ2-Leu,およびγ2-Val+δ1-Leuの組み合わせで標識したMSGでは,中間体法で得られるNOE強度の4倍の高感度で,よく分離したNOEシグナルが得られる(図2d, e).この結果,MSGのV118 γ1とV193 γ2-メチルの近傍にそれぞれV119 γ1およびL198 δ1-メチルが存在していることが明瞭となった.このように,メチル選択標識アミノ酸を用いる選択的標識技術は,高分子量タンパク質の構造情報の取得に有用である12).
最近になり,ILV生合成経路を遺伝子レベルで制御したILV要求性大腸菌変異株を用いて,標識ILVをより高い効率と選択性をもって目的タンパク質を標識する技術開発にも成功した.この結果,これまで報告されているさまざまな標識中間体を用いる手法に比べ,位置・立体選択的標識したILVを用いる手法の経済性,実用性がさらに高まると期待される13).
2)芳香環NMRシグナルの観測と構造情報の取得
高分子量タンパク質のNMR構造決定の精密化を達成するには,主鎖アミド(15NH)と側鎖メチル(13CH3)以外の原子に関する,できる限り多くの構造情報を取得することが望ましい.特に,芳香族アミノ酸の側鎖芳香環はかさ高く,またタンパク質内部の疎水性領域に数多く存在し,高次構造の形成や維持に深く関わるために,それらに関連したNMR構造情報は立体構造の精密化にとって重要である.従来の[13C, 15N]均一標識試料を用いる手法では,10~20 kDa程度のタンパク質においても,芳香環のNMRシグナルは複雑となるために,解析や帰属は困難な場合が多い.一方,SAIL法を利用することにより,NMRシグナルの測定や解析を妨げるさまざまな要因を取り除くことができるため,より高分子量のタンパク質でも芳香環NMRシグナルの構造情報の取得が可能となる14, 15).芳香族アミノ酸のみをSAIL標識したタンパク質試料の調製には,通常の大腸菌発現系が利用できるため実用性も高い.次に,転写因子Mybの高い相同性を持つ3ドメイン中,DNA結合に直接関与するR2–R3部位(Myb-R2R3)を例にとり,Trp残基のNMR情報の取得におけるSAIL法の利点を解説する.
Myb-R2R3は12 kDaの小さなタンパク質ではあるが,相同性の高い両ドメインに各6残基のTrp残基を含むため,均一標識体を用いる従来の方法では芳香環部分のスペクトルは複雑となり解析は困難である.一方,インドール環を構成する8個の炭素のうち,直接に結合していないδ1, ε3, η2の3か所のみを13CHとし,残りの水素をすべて重水素(D),炭素を核スピンのない12CのままにしたSAIL [δ1,ε3,η2]-Trp(図3a)では,芳香環のスピン系が著しく単純化される.このようなSAIL Trpを用いて選択標識したMyb-R2R3では,6残基のTrpのδ1, ε3, η2部位に由来する13CHシグナルがよく分離して観測された(図3b)16).本実験に用いたSAIL Trpは芳香環に加えて,脂肪族側鎖の炭素原子をすべて13C標識,さらにプロキラルβ-メチレン中のβ2(proR)水素のみを立体選択的に重水素標識してあるために,芳香環NMRシグナルをβ-13CH経由で帰属することが可能である.さらに,芳香環δ1-1H, ε3-1Hとβ3-1H,およびα-1Hとβ3-1Hとの間に観測されるNOE強度を比較することにより,側鎖立体配座(χ1, χ2)に関する構造情報が得られることも大きな利点である(図3c, d).
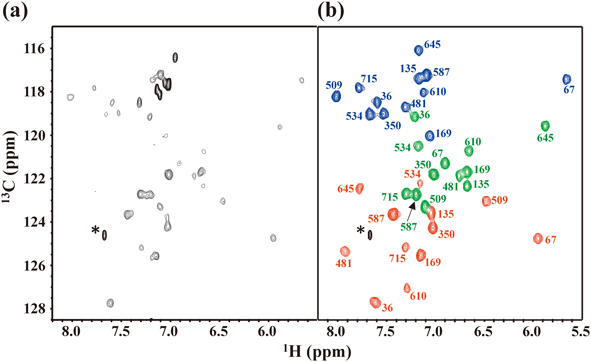
SAIL芳香族アミノ酸を用いて,どの程度までの高分子量タンパク質の芳香環領域のNMR構造情報が入手できるのであろうか? この疑問に答えるために,我々は高分子量NMR技術開発に広く利用されている82 kDaのリンゴ酸合成酵素(MSG)を対象にさまざまな検討を行った.このような高分子量のタンパク質では,NMRシグナルの線幅が拡がるために標識部位のアミノ酸以外をすべて重水素化しなくてはならない点が最初の課題となる.Mybタンパク質のように低分子量の場合には,周囲のアミノ酸が軽水素のままでも標識部位の芳香環シグナルの広幅化は無視できる.しかしながら,分子量が大きくなるにつれ周囲の軽水素(1H)核の影響(双極子緩和)や13C核の化学シフト異方性の効果が重なり,芳香環1H–13C NMRシグナルの観測が次第に困難となる.たとえば,炭素源として通常の(軽水素)グルコースにSAIL Trp(~10 mg/L)を添加した軽水最小培地を用いて,大腸菌高発現系により調製したMSGの1H–13C TROSY HSQCスペクトルでは,辛うじていくつかのδ1-, ε3-, η2-13CH相関シグナルが観測できるにすぎない(図4a).この程度のSAIL Trp添加量でも標識率は95%以上となるものの,周囲がすべて軽水素のために芳香環1H-NMRの線幅は著しく拡がりシグナルの観測を妨げるのである.一方,重水素化グルコースを用いた重水最小培地中にSAIL Trpを添加して調製したMSGの1H–13C TROSY HSQCスペクトルでは,13C核の化学シフト異方性緩和と直接に結合した1H核との双極子緩和が相殺されるため,よく分離した13CH相関シグナルが観測された(図4b).これら36個のシグナルは,MSGに12残基含まれるTrpのδ1-, ε3-, η2-13CHシグナルに由来し,全シグナルの帰属にも成功した(宮ノ入ら,投稿準備中).82 kDaのMSGに含まれるTrp以外の芳香族アミノ酸(Phe, Tyr, His)に関しても,同様な手法で芳香環13CHシグナルの観測・帰属が可能となる.このようにしてSAIL法を用いることにより,前項で紹介したメチルシグナルに加えて芳香環シグナルのNMR情報が得られ,高分子量タンパク質の溶液内での精密構造決定への道が切り開かれた.紙面の制限もあるために,SAIL法を用いた構造決定技術の最近の進歩に関しては別の機会に譲り,次節では芳香環反転運動を指標とするタンパク質動態について解説する.
1)芳香環反転現象を捉えるNMR手法
SAIL法がタンパク質NMR研究にもたらした最大の貢献は,各アミノ酸の安定同位体標識パターンの最適化により,難易度の高いタンパク質からも多様なNMR情報を合目的,かつ高感度に取得する可能性を大きく拡げたことにあろう.前節で紹介したSAIL[δ1,ε3,η2]-Trpの場合では芳香環の炭素を交互に13CH/12CD標識することにより,従来のNMR技術では困難であったインドール環NMRシグナルの高感度測定と配座解析への応用が可能となった.SAIL芳香族アミノ酸はこのような構造情報の取得に有用なだけではなく,芳香環部分の同位体標識パターンの特徴を活かしたタンパク質動態に関する研究にも大きく寄与する.
前節で述べたように,芳香族アミノ酸側鎖の立体配座は二つの二面角,χ1, χ2で定義される.このうち,χ2はCα–CβとCγ–Cδ間の二面角で定義され,Cβ–Cγ軸周りの立体配座に対応する.Trpのインドール環は非対称なχ2周りの回転構造を持っており,タンパク質中でχ2は+90°, 0°, −90°の3種類の異なった立体配座をとりうる.溶液内においては,結晶中と同様にTrp残基のχ2周りの立体配座はこれらの配座のいずれか,あるいはそれらの平衡混合物として存在すると考えられる.Phe/Tyr残基の芳香環のχ2周りの構造は対称性を持ち,結晶構造中ではχ2はおおむね±90°近辺の値をとるが,この二つの立体配座は定義により異なった符号を与えられものの,本質的には同一の配座である.χ2周りの回転構造が対称的であることは,Phe/Tyrの芳香環の反転前後の構造が完全に同一であるため,結晶構造解析によりχ2軸周りに静止しているのか,あるいは反転運動をしているのかを区別する手立てがないことを意味している.一方,溶液内では結晶中と同様χ2は±90°近辺の値に集中するものの,芳香環のχ2周りの動態をNMR法により捉えることができる.すなわち,Phe/Tyrの芳香環δ-, ε-シグナルは,Cβ–Cγ軸周りの反転運動が存在しなければ芳香環の両側面にある一対のδ1/δ2-,およびε1/ε2-CHが構造上非対称な局所的環境にとどまるために,それらは本質的に異なった化学シフトを持つ一対のNMRシグナルを与える.芳香環の周囲は疎水性アミノ酸側鎖に取り囲まれており,かさ高い芳香環の反転運動は抑制されると考えられるため,δ1/δ2-, ε1/ε2-CHが異なった化学シフトを示す残基が多数を占めるはずである.ところが,予想に反してタンパク質の1H-NMRスペクトルが測定可能となった1970年代当初から,タンパク質内部のPhe/Tyr残基においても一対のδ1/δ2-,およびε1/ε2-NMRシグナルを示すものはむしろ例外的であることが明らかとなった17–19).この事実は,芳香環のCβ-Cγ軸周りの速い反転運動によりδ1/δ2-,およびε1/ε2-NMRシグナルが平均化され単一のシグナルを与えるとしてのみ説明できる.結晶構造において,タンパク質内部の疎水性コア部分は揺らぎの少ない固い領域と思われていた当時,その領域に埋め込まれたPhe/Tyrの芳香環のようなかさ高い側鎖の速い反転運動を可能とする振幅の大きな揺らぎを持つことは予想もしなかった驚きであったであろう.繰り返しになるが,Phe/Tyrの芳香環の反転前後におけるタンパク質立体構造は不変であり,芳香環の反転現象はNMRによってのみ実験的に検証できる現象であることにもぜひ注目してほしい.芳香環はかさ高いといっても平面的であり,周囲を疎水性側鎖に緊密に取り囲まれて安定な位置を保ちうるが,その反転に際してはタンパク質内部に大きな空間が過渡的にでも生じる必要がある.このような過渡的空間の生成は,タンパク質内部に存在する協同的揺らぎに深く関わる動的現象として,現在に至るも多くの興味が集められている.歴史的には,1970年代末より精力的に行われたウシ膵臓トリプシン阻害剤(bovine pancreatic trypsin inhibitor:BPTI)に関するWüthrich–Wagnerグループによる一連の先駆的研究が広く知られているものの,その後のNMR研究は遅々として進まず今日に至っている19).このような遅れの主な理由は,観測される芳香環NMRシグナルが,反転運動速度に依存する線形(line-shape)変化を含めて,複雑となり定量的解析が困難となることによる.このような実験的障壁がSAIL法による芳香環スピン系の画期的な簡略化により大幅に取り除かれ,芳香環反転現象を利用したタンパク質の振幅の大きな揺らぎ(large-amplitude slow breathing motion:LASBM)の物理化学的研究,さらにはタンパク質の生物機能発現との関わりに向けた研究の新たな突破口が開かれた.次項では,この研究分野の嚆矢となったBPTIをモデルケースとして,SAIL法によって明らかにされる芳香環の反転運動とその構造生物学的意義に関して解説する.
2)トリプシン結合によるBPTIのLASBM揺らぎへの影響
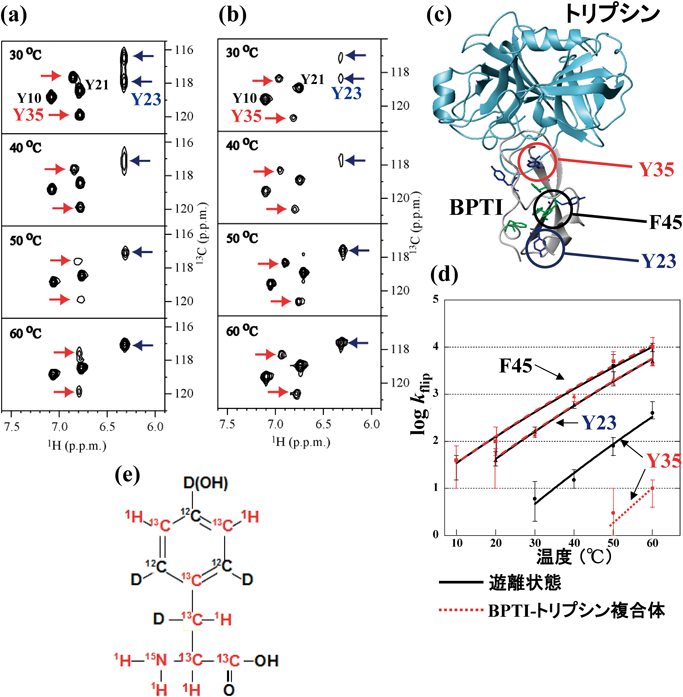
BPTIは58残基のアミノ酸からなるトリプシン阻害性の小タンパク質であり,高い熱的安定性を持つために,さまざまなNMR研究手法開発のモデルとして長い間利用されてきた19).我々は,芳香環反転運動のトリプシン結合による影響を調べるために,BPTI中に各4残基存在するPhe/Tyrを,ε-SAIL Phe/Tyr(図5e)により選択的標識したBPTIを調製した.このSAILアミノ酸の芳香環は,ε位のみが13C1H,残りはすべて12C,あるいは12CDとしてあるために,芳香環のNMRシグナルの線形解析には理想的な標識パターンを持つ.[ε-SAIL Phe/Tyr]BPTI, [ε-SAIL Phe/Tyr]BPTI-トリプシン複合体に関して,10~60°Cの範囲で測定した1H–13C相関NMRスペクトルから,複雑さを避けるため,図5a, bでは30~60°Cの範囲で測定したTyr 4残基のε-13C1Hシグナル部分のみを示した.なお,遊離状態のBPTIは低分子量タンパク質であるために通常のHSQC(図5a),トリプシン複合体中のBPTIでは分子量が増大するためTROSY HSQC(図5b)を測定した.この温度範囲では,Tyr23, Tyr35, Phe45残基以外の5残基のすべてのε1/ε2-CHは速い環反転運動により平均化され,各1本シグナルを生じる.したがって,図5a, b中,矢印のない2個の相関シグナルは平均化されたTyr10, Tyr21のε1/ε2-CHに由来する.一方,青矢印で示したTyr23シグナルは30°Cにおいて,ε1/ε2-1Hシフト差がε1/ε2-13Cシフト差に比べて小さいために1H軸(横軸)ではすでに平均化されており,13C軸(縦軸)に並んだシグナルとして観測される.しかしながら,30°Cでは13C軸の線幅の拡がりも顕著であり,40°C以上では平均化され1本のシグナルとなる.このように,Tyr23のε-CHシグナルの線形から,Tyr23の芳香環反転運動はトリプシンとの複合体形成により大きく影響されないことが明らかとなった.スペクトルは示さないが,Phe45のε1/ε2-CHのシグナルもTyr23と同様,複合体形成により反転運動は大きく影響されない.一方,赤矢印で示したTyr35は30°Cではε1/ε2位の13C-, 1H-化学シフト差がともに反転速度に比べて大きいために,40°Cまでは遊離状態,トリプシン結合状態のいずれにおいても類似したシグナルを与える.遊離状態では1H-化学シフトが小さいため50°C付近の1H軸ではすでに平均化され,化学シフト差の大きい13C軸では60°C付近になると13C軸での線幅の広がりが初めて明瞭となる(図5a).遊離BPTIに関する結果は,はるか以前に報告されたWüthrichらによる1H-NMRスペクトルの測定と基本的には一致する19).一方,SAIL法により初めて測定可能となったトリプシン複合体のスペクトルにおいては,Tyr35のε1/ε2位の化学シフト差は13C軸ではほぼ等しく,1H軸では遊離状態の2倍程度に拡大する.しかしながら,60°Cにおいても1H, 13C軸における環反転による線幅の拡がりは顕著ではない(図5b).
以上の推測をより定量的に検証するために,BPTIのTyr23, Tyr35, Phe45の遊離状態,およびトリプシン複合体中における環反転速度を,化学交換を測定するNMR実験(EXSY法)により測定したところ,Tyr35の芳香環の反転速度のみが複合体形成により顕著に低下することが明らかとなった(図5d).Tyr35残基の芳香環は,BPTI-トリプシン複合体の結晶構造(図5c)中では,直接にトリプシンと接触してはいないものの,複合体形成界面の近傍のBPTI内部に浅く埋め込まれた状態にある.遊離状態,複合体中におけるBPTIの結晶構造はきわめて類似していることから,BPTI-トリプシン相互作用は“硬い鍵と鍵穴の分子認識”の典型例として広く知られてきた.NMRにより明らかとなった,BPTIの活性部位ループ近傍に見いだされたLASBM揺らぎは,複合体形成後には大きく抑制されるものの,トリプシンとの複合体形成時には有利に働くと思われる(武田ら,投稿準備中).
Phe/Tyr残基の芳香環反転は,Cβ–Cγ単結合周囲のピコ秒以下で起こるきわめて速い回転振動現象であることを強調しておきたい.したがって,NMR法で測定される“速度”とは,実際にはかさ高い芳香環が反転するために必要な,ピコ秒程度の寿命を持つ160~170 Å3の過渡的空間が当該芳香環の周囲に生成する時間あたりの“頻度”を意味する20).このような大きな過渡的空間は分子表面近くであれば比較的頻繁に生じる可能性もあろうが,タンパク質内部等ではその生成頻度ははるかに低いと思われる.タンパク質主鎖や側鎖のランダムな揺らぎから偶然に大きな空間が生じ,その瞬間に芳香環が反転すると考えるのが妥当である.ε-SAIL Phe/Tyr,あるいはδ-SAIL Phe/Tyrにより選択標識したタンパク質を利用すれば,芳香環反転現象を通じたタンパク質のLASBM揺らぎとタンパク質間相互作用との関わりに関して新たな展望が得られる.次項では,このような観点から行ったタンパク質-薬剤相互作用界面におけるLASBM揺らぎに関する研究を紹介する.
3)タンパク質–薬剤相互作用界面の構造動態と薬剤認識
現在のタンパク質立体構造情報に基づく薬剤設計(structure based drug design)においては,薬剤–相互作用界面の立体構造は静止していることを前提としている.しかしながら,前項で紹介した芳香環の反転現象に反映されるタンパク質のLASBM揺らぎは,タンパク質内部の疎水性コア領域に限定されるわけではなく,薬剤–タンパク質相互作用界面においても同様に存在しうる.したがって,リガンドと結合することにより生物機能を発現するさまざまなタンパク質にとって,相互作用界面の構造動態と生物機能発現との関連は興味ある課題である.タンパク質–薬剤複合体界面の精密な立体構造情報に加え,さまざまな振幅とタイムスケールで揺らぐ構造動態を含めることにより,新たな薬剤設計指針にもつながる可能性がある.このような期待から,細胞内で免疫抑制剤FK506やラパマイシンと複合体を形成し,両薬剤に固有の生物活性の発現を促すFKBP12タンパク質をモデルとして,薬剤–タンパク質相互作用界面におけるLASBM揺らぎ変化を比較した21).
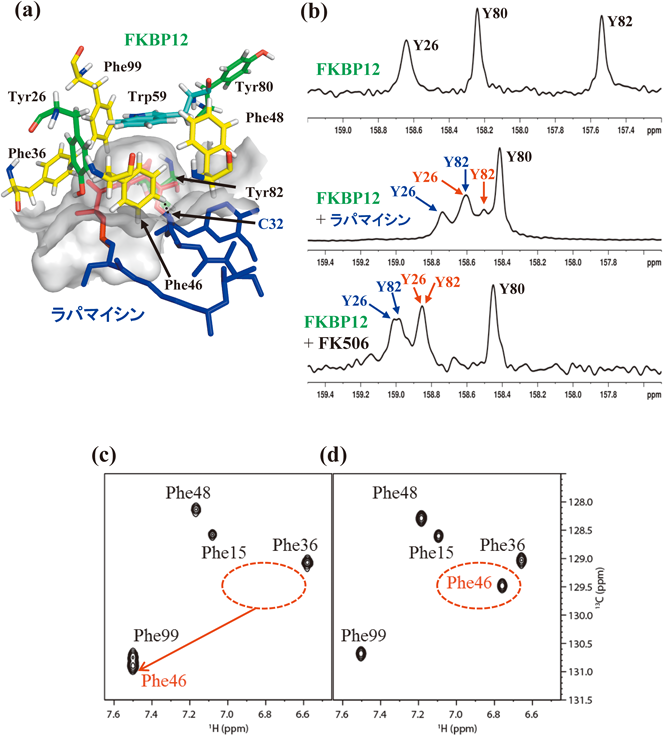
FK506とラパマイシンは,いずれも放線菌から見いだされた天然の環状マクロライド系免疫抑制剤であり,FKBP結合部位(FKBP binding domain)の化学構造は高い相同性を持つ.FK506(tacrolimus;タクロリムス)は細胞内に内在するFKBP12(FK506 binding protein)と複合体を形成することにより,マクロライド環におけるFKBP結合部位の逆側にある,固有のエフェクター結合部位(effector binding domain)と脱リン酸酵素カルシニューリン(calcineurin)との結合が著しく加速されることにより免疫作用が抑制される.一方,ラパマイシンにおいては,FKBP結合部位の構造はFK506とほぼ同一であるものの,エフェクター結合部位は大きく異なっている.この結果,ラパマイシンはFKBP12と複合体を形成することにより,mTOR(mammalian target of rapamycin)と特異的に結合し免疫作用を抑制する.このように,生物機能の発現機序は異なっているものの,両薬剤とFKBP12との複合体の高分解能結晶構造中の相互作用界面,すなわちFKBP12の薬剤結合部位,およびFK506,ラパマイシンのFKBP結合部位,に関して顕著な立体構造上の差異は見いだされない22, 23).両薬剤とFKBP12は解離定数がnM以下の強い複合体を形成することを考慮すると,複合体の相互作用界面は揺らぎのきわめて少ない構造をとっている可能性が高いようにも思える.このことを確認するために,我々はFKBP12の薬剤結合界面に芳香族アミノ酸5残基(Phe46, Phe48, Phe99, Tyr26, Trp59)からなる特徴的な芳香環クラスター構造が存在することに注目し,芳香環反転運動を指標として両複合体の相互作用界面のLASBM揺らぎを比較した.結晶中では両複合体のFKBP12の薬剤結合界面の芳香環クラスターの立体配置はきわめて類似しており,Trp59のインドール環の周囲をPhe46, Phe48, Phe99, Tyr26の芳香環がほぼ垂直に取り囲んだ半円筒形の疎水性ポケットを形成している.FKBP12-ラパマイシン複合体について図示したように,免疫抑制剤のFKBP結合部位(赤で表示)は疎水性ポケット内深くに挿入されている(図6a)22, 23).このように,FK506やラパマイシンが高い親和性を持ちFKBP12と強固に結合した状態においても,芳香環クラスター内のPheやTyr残基の芳香環反転が可能か否かは,タンパク質–薬剤複合体界面におけるLASBM揺らぎに関する新たな情報をもたらす.
4)FKBP12の薬剤結合界面の水素結合
薬剤結合に伴うFKBP12界面近傍の芳香族アミノ酸側鎖の水素結合に関する情報はTrp59の15NHε-化学シフト,およびTyr26の水酸基の1Hη-化学シフト,水素交換速度から得られる.Tyr26の水酸基の水素交換速度(pH 7.0, 30°C)は,我々が開発したTyr残基のζ13C-化学シフトへの水酸基の重水素化に伴う同位体効果を利用する手法により正確に測定した(図6b)24).すなわち,Tyr26の水酸基の水素交換速度は遊離のFKBP12では測定不能なほど速いものの,ラパマイシン複合体では約5.3 s−1に低下し,FK506複合体では<0.5 s−1とさらに1桁低下する.また,FK506複合体のTyr26の水酸基のOH化学シフト(9.75 ppm)はラパマイシン複合体(8.68 ppm)に比べて約1 ppm低磁場にシフトする.したがって,結晶構造から推定されていたFKBP12のAsp37側鎖カルボキシ基とTyr26の水酸基との水素結合は溶液中でも存在し,FK506複合体中ではラパマイシン複合体と比較して,この水素結合がより強固であることが示された.一方,Trp59のインドール環15NHεのNMRシグナルも有用な情報を与える.すなわち,通常は10 ppm付近にシグナルが観測されるインドール環1Hε/15Νεシグナルは,FK506複合体では5.25 ppm/120.4 ppm,ラパマイシン複合体では5.19 ppm/119.8 ppmに現れ,1Hシグナルは約5 ppmもの大きな高磁場シフトを示す.両複合体における化学シフト差は比較的小さいものの,この高磁場シフトはTrp59のインドール環NHがPhe99の芳香環とNH–π水素結合を形成しており,大きな環電流効果を受けていることを示している(図6a).
これらの水素結合は,いずれもFKBP12の分子内水素結合であるが,ζ-SAIL Phe標識FKBP12を用いて,FKBP12に5残基含まれるPheのζ-13CH NMRシグナルを両複合体間で比較したところ,予期せぬ重要な知見が得られた21).両複合体中のPhe残基は全般的にはきわめてよく似たζ-13CH化学シフトを持ち,Phe残基周囲の局所構造の高い相同性を示唆している.しかしながら,Phe46残基のζ-13CHシグナルに関しては,FK506複合体と比べラパマイシン複合体では約0.8 ppm(1H),約1.5 ppm(13C)もの大きな低磁場シフトを示す(図6c).このことから,ラパマイシン複合体の結晶構造中では2.5 Åの近距離にあるC32位のカルボニル酸素とPhe46のζ-水素間の水素結合が示唆された.このようなCH–O=C水素結合は結晶構造中では原子間距離や角度から,その存在がしばしば推定されてはいたが,NMRによる直接的証拠が得られた例はこれまでなかった.FK506ではこのカルボニル酸素の位置がメチル基となっているために水素結合を形成できず,Phe46のζ-CHに低磁場シフトは観測されない(図6d).FKBP12と薬剤間のこのわずか一つの分子間CH–O=C水素結合の有無が両複合体の結合界面の揺らぎに大きく影響する理由は後述する.
5)芳香環反転現象に現れるFKBP12–薬剤結合界面におけるLASBM揺らぎ
遊離状態のFKBP12においては,薬剤結合部位に存在するPhe46, Phe48, Phe99, Tyr26はタンパク質表面近くに存在しており,それらのδ1/δ2およびε1/ε2シグナルは速い反転運動により化学シフト位置,すなわち(δ1+δ2)/2および(ε1+ε2)/2,に平均化された各1本の13C/1Hシグナルとして観測される.一方,FK506,ラパマイシン複合体においては,芳香環クラスター内のPhe46, Phe48, Phe99, Tyr26のδ-, ε-CHシグナルは反転頻度に依存した明瞭な線形変化を示すが,クラスター外に存在する芳香環は測定条件下ではすべて平均化された1本のシグナルを与える.前述したように,芳香環の反転そのものはピコ秒程度の短時間で起こるCβ-Cγ軸周りの回転振動現象であり,芳香環周囲に反転に必要な160~170 Å3程度の空間が過渡的に生じる頻度が見かけの反転速度となる20).このような大きな空間が,はるかに頻度の高い主鎖や側鎖の振幅の小さなランダム運動(stochastic motion)からタンパク質内部のみならず薬剤結合界面においても偶発的に生じ,芳香環の反転に必要なピコ秒程度の間保持されることは,結晶構造からは予想できない事実であり,タンパク質による薬剤認識過程を理解する上で重要な知見となる.芳香環反転の前後でタンパク質の立体構造は完全に保たれるため,Phe/Tyrの芳香環反転現象はNMRによってのみ検出することができるタンパク質動態である点もあらためて強調したい.
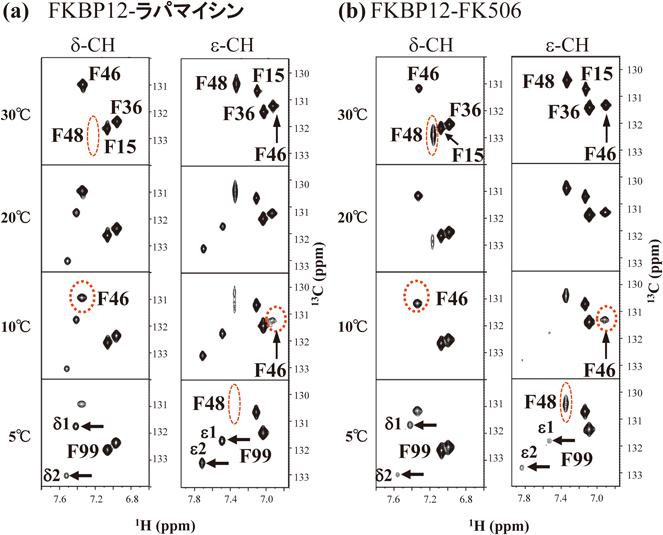
ラパマイシン複合体のδ, ε位の13C1Hスペクトル(図7)においては,Phe99のδ1/δ2, ε1/ε2シグナルは20°C以下で分裂し2本のシグナルとして現れる.一方,FK506複合体中ではδ, εシグナルはより低温の5°Cにおいて初めて分裂する.両複合体におけるPhe99のδ1/δ2, ε1/ε2シグナル間の化学シフト差はほぼ等しいことから,シグナルが分裂し始める温度がラパマイシン複合体に比べ,FK506複合体では著しく低いことはPhe99の反転速度がはるかに速いことを意味している.このように一定温度以下でδ1/δ2, ε1/ε2シグナルが分裂して観測できる場合には,反転速度をEXSY法により精密に決定することが可能であり,その結果5°CにおけるFK506複合体のPhe99の反転速度(kflip)はラパマイシン複合体に比べて約6倍速いことが見いだされた.さらに興味深いことに,両複合体界面に存在するTyr26の5°Cにおける環反転速度は,ラパマイシン複合体中で約50倍もFK506複合体中に比べて速い.このように,FKBP12と薬剤が結合する界面に隣接して存在するPhe99とTyr26の芳香環反転速度比が両複合体で大きく異なることが明らかとなった21).このことは,芳香環反転は複合体が解離し遊離状態となったFKBP12を経由せず,複合体上で生じることを意味している.それでは,なぜFKBP12の薬剤結合部位の芳香環クラスター中で隣接した位置にあるPhe99とTyr26は,両複合体の結晶構造中に顕著な差異が見いだされないにも関わらず,構造動態が大きく異なるのであろうか.この疑問は芳香環クラスター内のPhe46, Phe48残基の芳香環シグナルの線形の温度依存性から明らかとなる.
6)FKBP12-薬剤相互作用部位のLASBM揺らぎは薬剤構造の差異を鋭敏に反映する
Phe46, Phe48の芳香環δ1/δ2-, ε1/ε2-CHシグナルは,実験条件下において顕著な温度依存性を示すものの,Phe99やTyr26とは異なり分離したシグナルを与えないため,芳香環の反転速度を正確に測定することはできない(図7).また,Phe/Tyr残基の芳香環シグナルの線形は,反転速度とδ1/δ2-, ε1/ε2-CHシグナル間の化学シフト差の両者に依存するために,ある温度で見かけの線幅が両複合体で異なっていても,その差を直ちに反転速度と関連づけることはできない.しかしながら,前述したように速い反転速度により平均化され1本のシグナルとなった場合の化学シフトは,その温度におけるδ1/δ2-, ε1/ε2-CHシグナルの平均値となる.したがって,20°C以上の温度で観測されるPhe46, Phe48のε-CHの平均化されたシグナルの化学シフトが両複合体でほぼ等しいのは,Phe46, Phe48の芳香環ε1/ε2シグナルの化学シフト差が同程度であることを強く示唆している.ラパマイシン複合体中でのPhe48のε-CHシグナルは10°Cにおいて顕著に広幅化し,5°Cにおいて完全に消失するが,FK506複合体中では5°Cにおいてもシグナルが明瞭に観測される(図7a, b).このことは,FK506複合体中でのPhe48の環反転速度がラパマイシン複合体中に比べて格段に速いことを示している.同様に,両複合体のPhe46のδ-/ε-CHシグナルの線幅を詳しく比較すると,ラパマイシン複合体中でのPhe46の反転速度がわずかに遅いことが示された.これらのことから,Phe46のζ-水素がラパマイシンのC32位のカルボニル酸素と水素結合をしている影響は,Phe46自身の芳香環の反転速度よりも,むしろ隣接するPhe48残基の芳香環の反転速度に大きな影響を与えることが判明した.我々は,この理由をPhe48の芳香環が反転するためには,隣接するPhe46のζ-水素とラパマイシンのC32位のカルボニル酸素との間の水素結合が過渡的に切断されなければならないためであろうと推定した(図6a).FK506複合体には存在しえないζ-水素とカルボニル酸素との間の水素結合は,DFT計算から0.34 kcal/mol程度と推定される弱い水素結合ではあるが21),ラパマイシン複合体中でPhe46に隣接するPhe48の芳香環の反転速度を低下させるには十分であろう.
ラパマイシン複合体におけるPhe48の芳香環周囲の揺らぎがこの弱い水素結合で抑えられる影響は,FKBP12の芳香環クラスターの上蓋となるTrp59のインドール環を介して対岸のPhe99の芳香環周囲の揺らぎの低下を招く.この結果,ラパマイシン複合体中のPhe99の芳香環反転頻度は,FK506複合体の約1/6に低下する.FK506複合体におけるPhe99周囲のより大きい揺らぎは隣接するTyr26を押しやり,その結果,水酸基(OH)の低磁場シフトと水素交換速度の低下に反映されるようにTyr26の水酸基とAsp37側鎖カルボキシ基との水素結合は強まり,ひいてはTyr26の芳香環反転速度の著しい低下を招く.このように,FKBP12中のPhe46, Phe48, Trp59, Phe99, Tyr26から構成される芳香環クラスターの揺らぎは互いに密接に連携しており,Phe46とラパマイシンの間のわずか一つの水素結合の影響は薬剤相互作用部位全体へと伝播すると思われる21).ラパマイシンはFKBP12と複合体を形成することにより,mTORのFRB(FKBP-rapamycin-binding)ドメインと強く結合し,mTORの機能を阻害し免疫作用を抑制することは既に述べたが,その理由に関しては必ずしも明確ではない25, 26).FKBP12-ラパマイシン-FRB複合体(ternary complex)の結晶構造ではわずかな直接的相互作用がFRBとFKBP12間に観測されるものの,ラパマイシンのFRB結合ドメイン(エフェクター結合部位)の構造はFKBP12-ラパマイシン複合体(binary complex)とほぼ等しい27).このように,結晶構造を詳細に調べても何故FKBP12と結合することによりラパマイシンとmTORとの結合が遊離状態の2000倍も強まるのかは解明することはできない26).
以上に述べたように,FKBP12のラパマイシン,およびFK506との複合体形成部位を形成する芳香環クラスターは,両複合体の結晶構造をみる限りきわめて類似しているものの,クラスター内に存在する芳香環反転現象をSAIL法により詳細に検討したところ,両複合体の複合体形成界面のLASBMには特徴的な差異が見いだされた.このような複合体形成界面の動態変化が,FKBP12-ラパマイシン複合体にのみ存在するC32位のカルボニル基とFKBP12のPhe46のζ–H間の水素結合に起因するならば,ラパマイシンのFKBP結合部位における構造上の微細な違いにより引き起こされる複合体界面のLASBM変化が,mTORとの相互作用に影響を与えるか否かを実験的に検証することはきわめて興味深い.たとえば,ラパマイシンのC32位のカルボニル基をメチレン基に置換できたとすれば,FKBP12–薬剤結合部位のLASBM変化がエフェクター結合部位の動態変化を介してmTORとの相互作用に影響が見られるであろうか? もし,タンパク質の薬剤結合部位の構造動態を薬剤設計により制御できるならば,SAIL法により明らかとなるタンパク質–薬剤複合体界面の構造動態は新たな薬剤設計指標となる可能性がある.