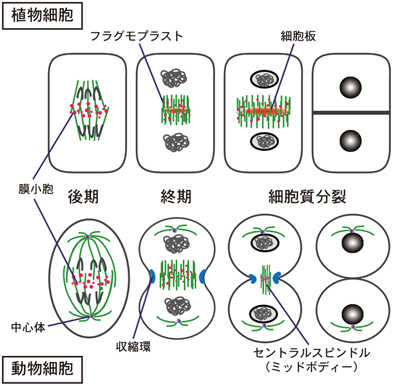
細胞分裂において,細胞質分裂は遺伝情報を正確に娘細胞に伝達する上で最後の重要なステップである.染色体の複製から分離までの過程は,種々の生物間で大部分が保存されているが,細胞質分裂の過程は各生物間で異なっているようにみえる.たとえば,動物細胞では,アクチンとミオシンを主成分とする収縮環と,微小管を主成分とする細細胞質分裂装置,セントラルスピンドル(中央紡錘体)に依存して外側から内側へ向かって細胞質分裂が進行するのに対し,植物細胞では収縮環は存在せず,二つに分離した娘細胞の間に形成される微小管を主成分とする細胞質分裂装置であるフラグモプラスト(隔膜形成体)に依存して形成される細胞板が内側から外側へ向かって拡大することにより細胞質分裂が進行する(図1).このような違いは,それぞれの細胞の独自の構造や機能に依存していると考えられる.植物細胞は,細胞膜の外側に強固な細胞壁を有しているが,細胞壁の合成を伴いながら分裂を進行させる必要があることを考えると,植物の内側から外側に向かう分裂様式は,合理的な方法である.
植物の細胞質分裂は,主に次の三つの段階,1)細胞周期M期の後期に開始されるフラグモプラストの形成,2)フラグモプラストの拡大成長,3)細胞板の形成,を経て実行される.フラグモプラストは,動物のセントラルスピンドルと同様,重合,脱重合の起こりやすいプラス端と,より安定なマイナス端という極性を持った二つの微小管の束により形成されており,二つの微小管束は互いにプラス端側で向かいあっている(図2A).初期フラグモプラストは,娘染色体の間に存在する残存する紡錘体微小管,もしくは新たに生成された微小管が,プラス端で互いに組み合わされ,それが束になり,内側まで微小管が詰まった樽状の構造体として作られる.このようなフラグモプラストの中でいったん細胞板が形成されると,フラグモプラストは細胞板を取り巻くようなリング状の構造となり,細胞板の拡大を伴いながら遠心的に拡大し親細胞壁まで到達することで細胞質分裂を完了させる(図1).細胞における初期フラグモプラストの位置の決定,拡大伸長方向の決定,親細胞壁内側の接続部位(到達部位)の決定は分裂面の位置決めと深く関わり,その仕組みの解明は多細胞生物の発生を理解する上で重要であるが,まだほとんどわかっていない.
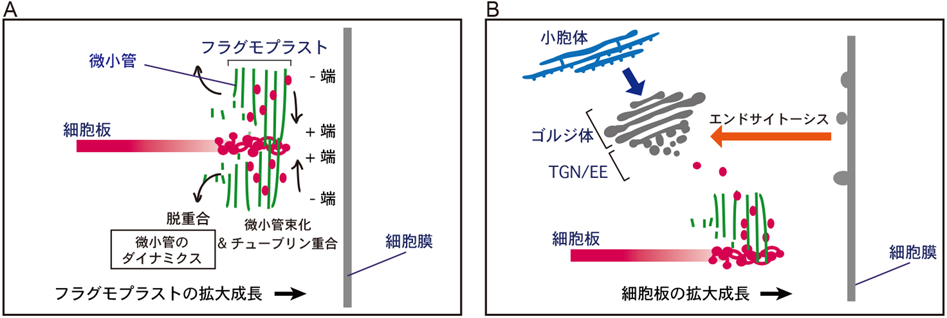
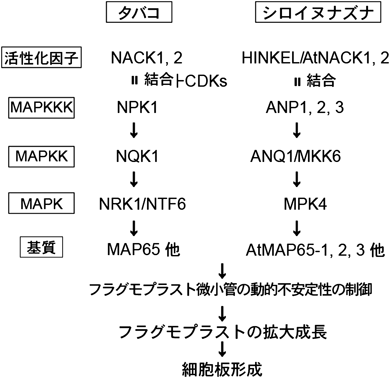
フラグモプラストの拡大成長は,微小管束の内側での脱重合と外側でのチューブリンの重合によって保証されていると考えられている(図2A).このような微小管の脱重合とチューブリンの重合による微小管の変動は微小管のダイナミクスと呼ばれている.フラグモプラスト微小管は,細胞板の材料を含むゴルジ,細胞膜由来の小胞を運ぶレールの役割をしていると考えられ,細胞板の形成と成長はフラグモプラストの形成と遠心的拡大に依存していると言い換えることができる.つまり,上記した2)と3)の過程は密接に関係していると考えられるが,実際に,さまざまな阻害剤を用いた実験から,この二つの過程は高度に共役して進行していることが指摘されている.たとえば,ゴルジ体を介する膜交通システムの阻害剤であるブレフェルディンA(BFA)で処理すると,細胞板への膜成分の集積は阻害されるが,同時にフラグモプラストの拡大も阻害される1).この結果は,細胞板形成がある程度進行した後に,フラグモプラストが拡大するような調節機構が働いている可能性を示している.我々は,植物の細胞質分裂に伴うフラグモプラスト微小管の動態の制御,つまり,上記した細胞質分裂の三つの過程のうち,主に2)の過程には特異的なMAPキナーゼカスケードが関与していることを明らかにしてきた(図3)2–10).ここでは,まずそのカスケードによる制御メカニズムを中心に,植物の細胞質分裂の分子機構について説明した後に,細胞板形成を担う膜交通システムについて解説する.研究に使った主な生物材料は,タバコ培養細胞BY-2およびシロイヌナズナである.
2. 植物の細胞質分裂を制御するMAPキナーゼカスケード
1)植物のMAPキナーゼカスケード
動物細胞では,細胞質分裂を含むさまざまな分裂期のイベントを協調的に進行させる分裂期キナーゼと呼ばれる制御因子が多数同定されている11).しかし,植物ではそのオーソログが存在しないものも多く,植物は独自の制御系を進化させてきたと考えられる.植物の細胞質分裂に関わる制御因子として,現在最も解析が進んでいるのは細胞質分裂時特異的に活性化するMAPキナーゼカスケードである.
MAPキナーゼカスケードは,真核生物に保存されたシグナル伝達経路で,MAPキナーゼ(mitogen-activated protein kinase:MAPK),上流のMAPキナーゼキナーゼ(MAPKK),さらに上流のMAPキナーゼキナーゼキナーゼ(MAPKKK)とともにカスケードを構成しており,連続した三つの酵素の組み合わせにより,種々の細胞内,細胞外シグナルを特異的な経路に伝達し,適切な細胞応答を誘導する.このカスケードの活性化は,MAPKKKの活性化から始まる.活性化されたMAPKKKは,下流のMAPKKに保存されるS/T-x3-5-S/Tモチーフ中の二つのセリンまたは,トレオニンをリン酸化することによりMAPKKを活性化する.活性化型MAPKKは,下流のMAPKの活性化部位に保存されるT-x-Yモチーフ中のトレオニンとチロシンをリン酸化することによりMAPKを活性化する.活性化されたMAPKは,標的分子中に含まれるP-x-S/T-PもしくはS/T-P配列中のセリンまたは,トレオニンをリン酸化し,標的分子の活性を制御している.植物ゲノム中にも,動物と同様に多数のMAPキナーゼカスケードの構成因子が存在することがゲノムプロジェクトの完了とともに明らかになっており,シロイヌナズナは20個のMAPK, 10個のMAPKK,そして約60個のMAPKKK遺伝子12)を,イネは15個のMAPKと8個のMAPKK遺伝子を,ポプラは21個のMAPKと11個のMAPKK遺伝子を持つことが報告されている13).
MAPキナーゼカスケードの活性化に関わる最初のメンバーであるMAPKKKの遺伝子は,大きな遺伝子ファミリーを形成しており,動物のMAPKKKとの相同性から,MEKK様キナーゼとRaf様キナーゼの大きく二つのサブグループに分類される.MEKK様キナーゼは多くの場合,MAPKKKとして機能することがすでに証明されており,シロイヌナズナでは,後述する細胞質分裂の制御に関わるANP1, ANP2, ANP3,病害抵抗性に関わるMEKK1,そして胚発生において重要な機能を持つYODAがこのグループに分類される.Raf様MAPKKKは今のところ,MAPKKKとして機能するかどうかの実験的証明がなされていないものが多い.MAPKKとMAPKは,それぞれ動物や酵母とアミノ酸配列の相同性も高く,機能的にも類似した性質を有している.ただし,動物,酵母のMAPKが三つのサブファミリー(ERK, JNK/SAPK, Hog/p38)に分類されるのに対し,植物のMAPKはアミノ酸の相同性による分類ではすべてERKファミリーに属し,JNK/SAPK, Hog/p38サブファミリーと相同性の高いMAPKは見つかっていない12).
2)植物の細胞質分裂を制御するNPK1-NACK1複合体
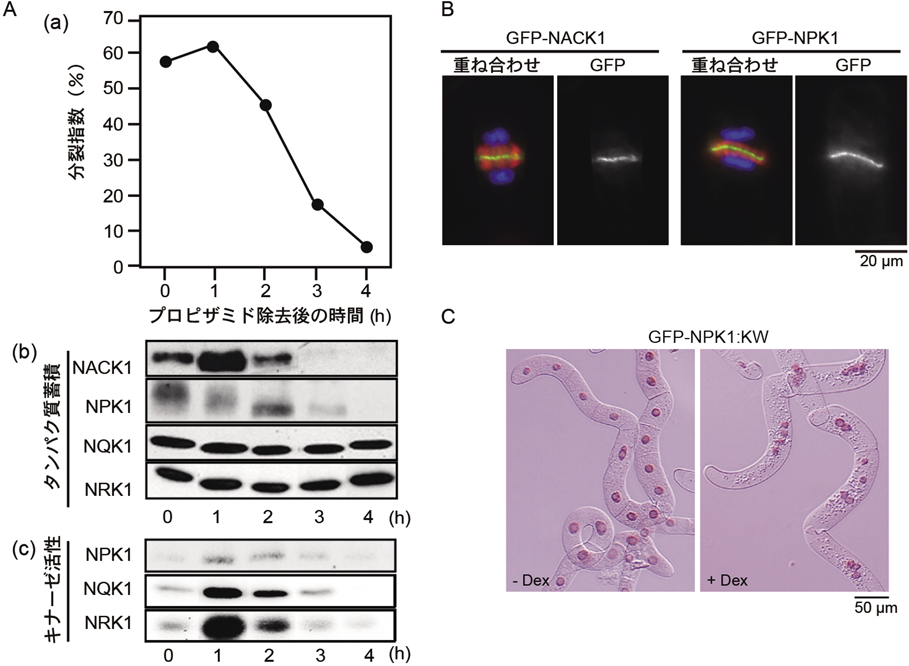
NPK1(nucleus-and phragmoplast-localized protein kinase 1)は,植物で同定された最初のMAPKKKであり,タバコ培養細胞BY-2において対数増殖期に特異的に発現するプロテインキナーゼとして同定された.BY-2細胞は増殖が早く(対数増殖期では細胞周期は約12時間),DNA合成阻害剤アフィディコリンと微小管脱重合剤プロピザミドを組み合わせることにより,70%以上の細胞で同調培養が可能な細胞株である.ここで紹介する研究は,特定しない限りこの細胞株を用いて行われた.同調培養したタバコ培養細胞において,NPK1タンパク質は,S期からM期終了までの間に蓄積するが,その活性はM期後半に限定されている(図4A).NPK1は細胞周期において核膜が崩壊するまでは核に局在しているが,核膜崩壊後から中期までは細胞質中に散在し,染色体が分離し,フラグモプラストが形成されると,フラグモプラストの赤道面にその局在を変化させる5).NPK1の活性は細胞質分裂の時期にピークを迎えるが,このとき,NPK1は拡大するフラグモプラストの赤道面(+端側)にリング状の局在を維持し続ける(図4B).我々が用いた培養条件では,個々のBY-2細胞で形成されたフラグモプラストは40分から1時間で親細胞壁に到達する.
BY-2細胞においてドミナントネガティブ型(キナーゼ不活性型)のNPK1を過剰発現すると,フラグモプラストの拡大成長が阻害され,不完全な細胞板を持つ多核化した細胞が生じる(図4C)5).この表現型は微小管安定化剤であるタキソールで処理したときの表現型と似ていたことから14),NPK1はフラグモプラストの拡大成長の基盤となっている微小管の再構築過程に関与していることが予想された.NPK1活性が細胞質分裂時に一過的に上昇すること,NPK1がフラグモプラスト赤道面(微小管のプラス端)に局在することは,この予想を完全に支持する.シロイヌナズナには,NPK1ホモログが三つ存在するが(ANP1, ANP2, ANP3),これらもタバコと同様に細胞質分裂に関与している.anp2 anp3二重変異体は,不完全な細胞板を持つ多核化細胞を有する細胞質分裂異常を示し,さらに三重変異体は配偶体致死となった15).このようにNPK1 MAPKKKファミリーの活性は,植物の細胞質分裂の進行に必須であることが明らかとなった.
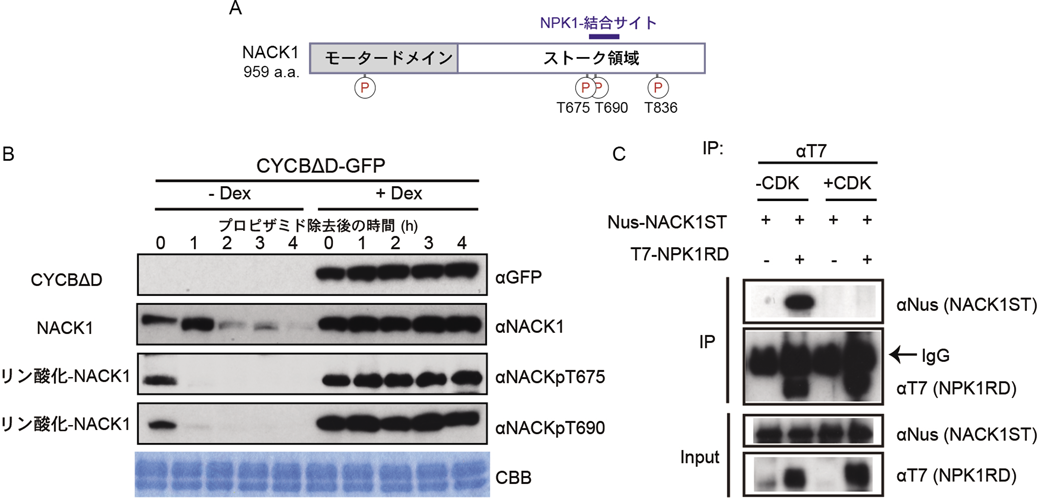
NPK1は,C末端の制御領域を除去すると恒常的に活性化することが明らかとなっていたことから,全長は不活性型であり,このMAPKKKの活性化には何らかの制御因子が関与していることが示唆されていた.そこで,酵母の接合因子応答性MAPキナーゼカスケードを利用した機能相補スクリーニングが行われ,NPK1 MAPKKKの活性化因子として二つのキネシン様タンパク質の遺伝子cDNA(NACK1とNACK2)が単離された6).両遺伝子はN末端側に微小管モータードメイン,C末端側にはコイルドコイル構造に富んだストーク領域を持つ典型的なキネシン様タンパク質をコードしていた.NACK1遺伝子およびNACK1タンパク質はM期特異的に蓄積を示し,その蓄積パターンはNPK1の活性化のパターンと一致している(図4A).タバコ培養細胞における細胞内局在もNPK1と一致しており,細胞質分裂時にはフラグモプラストの赤道面に局在する(図4B)6, 16).このタンパク質はNPK1と直接結合し,この結合がNPK1の活性化を誘導することがin vitroおよびin vivoで示されている.モータードメインを欠損させたNACK1をタバコ培養細胞において過剰発現させると,NPK1のフラグモプラスト赤道面への局在が阻害されると同時に,フラグモプラストの拡大成長が停止し,多核化した細胞が出現する.これらの結果から,NACK1は細胞質分裂において,NPK1の活性化と同時にNPK1をフラグモプラストの赤道面へリクルートする因子であると考えられている6).
シロイヌナズナにおけるNACK1のオーソログであるAtNACK1/HINKEL(HIK)とTETRASPORE(TES)/STUD/AtNACK2もまた,ANPの活性化を介して細胞質分裂の進行に関与している1, 2, 6, 7, 9, 10, 17).atnack1/hinkel変異体とtes/stud/atnack2変異体は,それぞれ体細胞および雄性配偶子において細胞質分裂に異常が生じる1, 2, 6, 7, 9).これらの結果から,シロイヌナズナにおいてもそれぞれのNACKタンパク質は細胞質分裂に寄与していることがわかった.さらに二重変異体は,雄性配偶子,雌性配偶子のいずれにおいても細胞質分裂異常になり,致死となることから,これら二つの遺伝子は重複した機能を持ち,細胞質分裂に必須の因子であることが示された10).NACKタンパク質は,イネやヒメツリガネゴケにおいても細胞質分裂に関与していることから18, 19),本タンパク質は植物において保存された細胞質分裂の鍵因子として機能しているのであろう.
3)NPK1-NACK1複合体の下流で働くMAPKKとMAPK
NPK1 MAPKKKの下流に位置するNQK1 MAPKKは,酵母の浸透圧ストレス下で活性化されるMAPキナーゼカスケードの変異体を用いた相補スクリーニングにより単離された8).また,その下流のNRK1 MAPKはNQK1 MAPKKを用いた酵母のツーハイブリッド法により単離された8).NRK1は,M期の後半に活性化され,フラグモプラストの赤道面に局在することが報告されているNTF6 MAPKと同一であった20).NQK1はNPK1と,NRK1はNQK1とin vitroで直接結合することから,NACK1, NPK1, NQK1, NRK1は全体として複合体を形成していると考えられる.また,NPK1, NQK1, NRK1のそれぞれは,上流の因子が下流の因子をリン酸化しキナーゼ活性を上昇させる8).NQK1とNRK1のタンパク質量は細胞周期を通して変化がないが,その活性はM期の後半に上昇する(図4A).またこれら二つの因子は,細胞質分裂時には,NACK1とNPK1と同様にフラグモプラストの赤道面に局在することも示されている17, 20).
シロイヌナズナの変異体の解析から,MKK6およびMPK4が,タバコのNQK1とNRK1のホモログであることが明らかとなっている.これら因子の変異体は,いずれも細胞質分裂異常を示す細胞を有し,矮性の表現型を示す8, 17, 21, 22).MKK6はAtNACK1もしくはAtNACK2存在下でANP MAPKKK(ANP1, ANP3)によりリン酸化され活性化される.活性化型MKK6は,MPK4と同じグループBに分類されているMAPK(MPK4, MPK5, MPK11, MPK12, MPK13)のうち,in vitroではMPK4のみをリン酸化して活性化する17).加えて,MKK6とMPK4は,シロイヌナズナ個体において,細胞質分裂時に細胞板形成部位に局在する17, 21, 22).このように,タバコにおいてはNPK1 MAPKKKとNQK1 MAPKKとNRK1 MAPKが,シロイヌナズナにおいてはANP MAPKKKとMKK6 MAPKKとMPK4 MAPKが,それぞれ固有のリン酸化カスケードを構成し,細胞質分裂時に細胞板の拡大成長を制御していることがわかった(図3).このカスケードは,キネシン様タンパク質であるNACKがMAPKKKに結合することにより活性化されることから,経路全体をNACK-PQR経路と名づけている8).
3. NACK-PQR経路の標的分子と細胞質分裂の制御
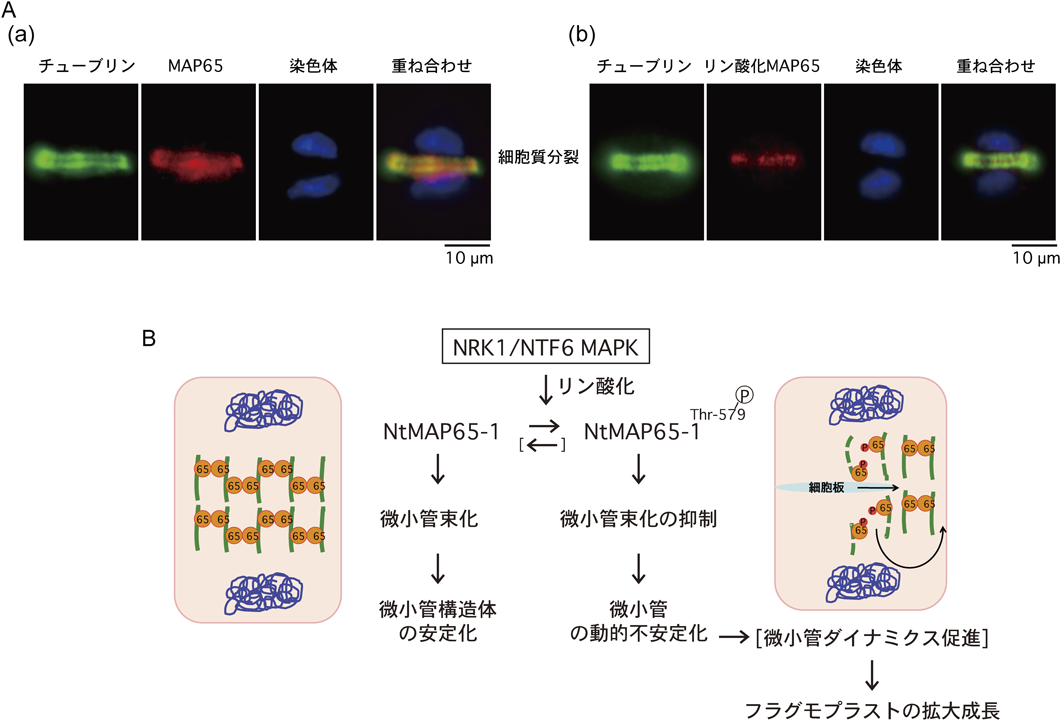
NACK-PQR経路の各構成因子は,いずれもフラグモプラスト微小管の赤道面(プラス端側)に局在することから,この経路は,フラグモプラスト微小管の動態を制御していることが予想された.そこで,我々は,タバコの微小管結合タンパク質(microtubule-associated protein:MAP)に着目し,NRK1 MAPKの標的タンパク質の探索を行った.その結果,タバコBY-2細胞より調製したMAP画分中に,NRK1によりリン酸化されるタンパク質を複数見いだした.その中の一つは,MAP65ファミリーに属するNtMAP65-1であった.このファミリーは植物(MAP65),酵母(Ase1p),線虫(SPD1),ショウジョウバエ(Feo)から哺乳類(PRC1)に至るまで広く保存されている.いずれの生物においても,このファミリーのタンパク質は細胞質分裂装置に局在すること,変異体において細胞質分裂の異常が観察されることなどから,細胞質分裂における役割が報告されているが,それによる制御の仕組みについては不明であった23–28).MAP65ファミリーのタンパク質は,いずれも微小管結合能および微小管束化能を持つ.MAP65によって形成された束化微小管は,微小管脱重合剤や,低温ストレスのような環境ストレスにより誘導される微小管の脱重合に対して抵抗性が増すことが知られている29–32).MAP65の微小管束化活性は,動物のセントラルスピンドルや植物のフラグモプラストのような微小管構造を維持するのに必要であると考えられている.
タバコのNtMAP65-1はMAPKによるリン酸化部位を1か所持つ.タバコ培養細胞BY-2において,MAPKによるNtMAP65-1のリン酸化レベルはM期中期以降に最大となり,NRK1/NTF6の活性化パターンと対応していた.この時期に,MAP65はフラグモプラスト全体に局在していたが,リン酸化抗体を用いて,MAPKによりリン酸化されたNtMAP65-1の局在を解析したところ,NRK1/NTF6 MAPKと同様に,フラグモプラストの赤道面に局在していた(図5A).つまり,リン酸化型のNtMAP65-1はフラグモプラスト上でプラス端に存在していた.このリン酸化はMAP65の機能にどのような影響を与えるであろうか? NRK1によるリン酸化は,in vitroでNtMAP65-1の微小管束化活性を抑制する.さらに,NRK1リン酸化部位のセリンをアラニンに置換したMAPK非リン酸化型NtMAP65-1をタバコ培養細胞で過剰発現させると,微小管束の安定性が上昇し,微小管脱重合試薬に対する抵抗性が増大すると同時に,フラグモプラストの拡大成長が著しく遅延した33).これらの結果から,我々は,細胞板の周縁に位置するフラグモプラストの赤道面では,NRK1 MAPKが,NtMAP65-1タンパク質をリン酸化しフラグモプラストの束化を局所的に緩め,微小管の不安定性を亢進することにより,フラグモプラストの拡大成長を促進するというモデルを提唱している(図5B)33).
シロイヌナズナには九つのMAP65ファミリーのメンバーが存在するが,そのうち,MAP65-3の変異体は,細胞質分裂不全を示すことがわかっている26, 34).また,AtMAP65-1はタバコのNtMAP65-1と同様にフラグモプラストに局在し,微小管束化活性がリン酸化により抑制されることが報告された35).map65-1変異体や,map65-1 map65-2二重変異体では細胞質分裂の異常は観察されないが,map65-1 map65-3,およびmap65-2 map65-3二重変異体では,map65-3変異体よりも強い細胞質分裂異常が観察されることから,これらは細胞質分裂において重複した機能を有していると考えられている36).これらは,in vitroでMPK4(NRK1のシロイヌナズナホモログ)によってリン酸化されることも示されているが,そのリン酸化の効果については未解明である.動物細胞では,MAP65ファミリーのタンパク質の微小管結合活性がCDK(cyclin-dependent kinase)により制御されていることが報告されているが,このような違いが,細胞質分裂における微小管機能の違いとしてどのような意味を持っているのかも興味深く,今後の課題であろう.
NACK-PQR経路の活性の維持に関しては,フラグモプラスト微小管との関連においてもう一つ特徴がある.すでに述べたように,この経路はフラグモプラスト微小管のダイナミクスを制御することによりフラグモプラストの拡大成長を促進しているが,微小管自身が経路の活性化や維持に必要であることがわかっている.具体的には,プロピザミド添加により拡大成長しているフラグモプラスト微小管の脱重合を誘導すると,カスケードの三つのキナーゼの活性が消失するのである8).このように,この経路の活性は微小管の脱重合によりフィードバック阻害を受けるが,その仕組みについては不明である.
細胞質分裂を含む分裂期の各過程の進行は,NACK-PQR経路の活性化も含めて厳密に制御されている.NACK-PQR経路の特異的活性化は,活性化因子であるNACK1遺伝子のM期特異的な転写と,細胞周期がM期中期から後期に進行したときにのみNACK1がNPK1 MAPKKKに結合することに依存している.NACK-PQR経路が正確なタイミングで細胞質分裂を実行するために重要なこの二つの制御は,いずれも細胞周期進行の中心的な制御因子であるCDKキナーゼ/サイクリン複合体によるものであることが示されている.ここでは,CDKによるNACK1に関する二つの制御システムについて解説する.
1)CDKによるNACK1のM期特異的転写制御の仕組み
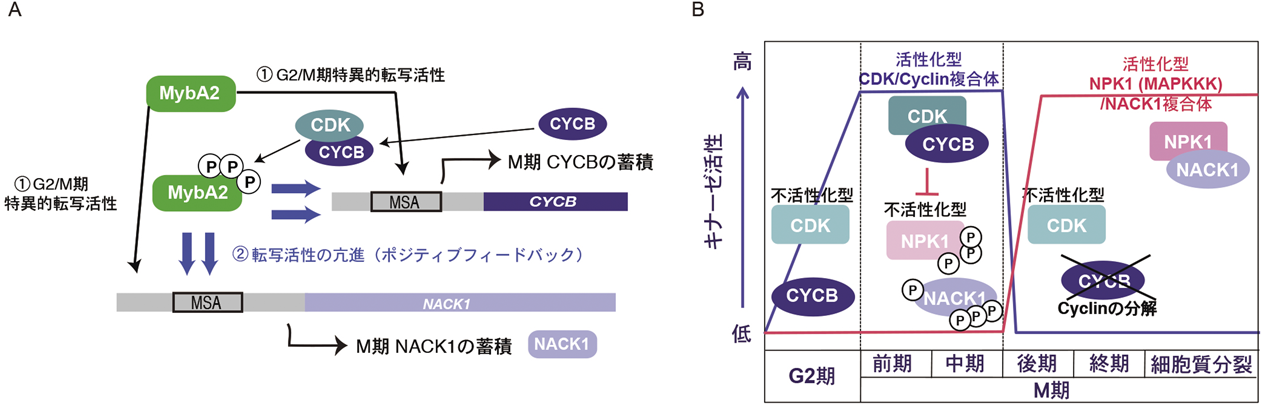
NACK1のmRNAとタンパク質の蓄積はG2/M移行期に観察され始める.そして,NACK1タンパク質はM期の間蓄積し,細胞質分裂の終了と同時に分解される6).NPK1遺伝子mRNAとタンパク質の蓄積はS期から細胞質分裂の終了まで,NQK1およびNRK1遺伝子mRNAとタンパク質は細胞周期を通じて存在している(図4A-b).したがって,NACK-PQR経路の細胞質分裂時特異的な活性化の仕組みの一つはNACK1の転写制御に依存していると考えられる.G2/M期に転写されるCYCB(Cyclin B)遺伝子やNACK1遺伝子を含むM期特異的遺伝子は,MSA(mitosis-specific activator)エレメントと呼ばれる共通のシス配列を持つことが知られている.MSAエレメントは,G2/M移行期に転写される多くの遺伝子のプロモーター領域に存在し,三つのMybドメインを持つMyb転写因子(R1R2R3-Myb)によって認識される転写のエンハンサーとして機能する37–41).タバコのR1R2R3-MybであるNtMybA1とNtMybA2遺伝子は,自身もG2/M移行期に特異的に転写され,蓄積したMybタンパク質がCYCBとNACK1遺伝子のプロモーター領域に存在するMSAエレメントの機能を介してそれらの遺伝子の転写を誘導する42).
NtMybA2の転写活性化能はC末端側の制御ドメインによって抑制されているが,この抑制効果は,CDKによるNtMybA2のリン酸化によって解除される.NtMybA2の制御ドメインにはCDKによってリン酸化されるセリンとトレオニン残基が20個存在しているが,この残基がリン酸化されることにより,その転写活性は上昇し,その活性に依存してNACK1およびCYCBの転写レベルが上昇する(図7A参照)43).M期CDKの活性化にはCYCBが必要なので,NtMybA2によって発現誘導されたCYCBはCDKを活性化して,NtMybA2をリン酸化し,さらにその転写活性化能を上昇させる.このようなR1R2R3-Myb転写因子の転写と活性化に関するポジティブフィードバックループによりR1R2R3-Myb転写因子の蓄積が促進され,その結果,M期に必要なCYCBやNACK1のような遺伝子の迅速な転写とタンパク質の蓄積が保証されている(図7A).
2)細胞質分裂開始前のCDKによるNACK-PQR経路の抑制メカニズム
NACK-PQR経路の活性化は細胞質分裂時に限定されているが,先にも述べたとおり,NACK1およびNPK1はM期の中期以前からは細胞内に蓄積している.つまり,この経路の細胞質分裂時特異的な活性化は遺伝子の転写翻訳時期だけでは説明できず,何らかの翻訳後修飾が関与していることが示唆されていた.NACK-PQR経路の活性化はNACK1とNPK1の結合に依存しているが,この間の研究により,CDKがこの結合を阻害していることが明らかになった.
NACK1とNPK1にはCDKによるリン酸化保存配列がそれぞれ,4個(図6A)もしくは3個存在しているが,これらのアミノ酸残基はin vitroでCDKによりリン酸化される16).同調化したタバコ培養細胞を用いてこれらリン酸化候補部位の生体内でのリン酸化レベルを検出したところ,NACK1, NPK1ともにM期の前半に特異的にリン酸化されていることが示された.両タンパク質がリン酸化されている時期はCDK活性が高い時期と一致している.そして,リン酸化レベルが低下する時期にNPK1キナーゼの活性が上昇することから,両タンパク質のリン酸化は,NPK1の活性化を負に制御していることが予想された.CDKは,M期中期を通過すると活性調節因子であるCYCBが分解されることにより活性が急激に低下するが,CYCBのデストラクションボックス(D box)に変異を入れたCYCB-ΔDを培養細胞において過剰発現させると,中期以降もCYCBが分解されずCDK活性が高く保たれ,後期の開始が遅延する44).この実験系を用いて,NACK1とNPK1のリン酸化状態を解析したところ,M期前半におけるリン酸化は,CDK活性によることが明らかになった(図6B)16).両タンパク質のリン酸化は,NPK1とNACK1の直接結合をin vitro, in vivoの両方で阻害することも示された(図6C).つまり,CDKの活性が高いM期中期までは,NACK1とNPK1はリン酸化され,その結合が阻害されていることがわかった.中期以降になるとCDK活性が低下し,何らかの仕組みで脱リン酸化が起こり,NPK1とNACK1が結合してNPK1キナーゼが活性化されると推察される(図7B).このように,NACK-PQR経路の活性化は,細胞周期を制御する中心的制御系であるCDK/サイクリン複合体の制御下で制御されている.今後は,NACK-PQR経路の活性化に関わると期待される脱リン酸化酵素の同定が重要であろう.
フラグモプラスト微小管は,極性に関する特徴から,紡錘体微小管が起源であるという可能性が示唆されていたが45, 46),フラグモプラスト形成には,新たな微小管重合も伴うことが報告されている47).現在までのところ,初期フラグモプラスト形成の過程は明らかになっていないが,フラグモプラストの拡大に関わる分子メカニズムについては,オーグミンとγチューブリンのフラグモプラストでの役割が示されたことにより,大きく理解が進んだ.
オーグミン-γチューブリン複合体は,ショウジョウバエの紡錘体形成において,微小管の重合核として機能することが報告された48).この複合体が最近,ヒメツリガネゴケにおいて,拡大中のフラグモプラストでの微小管生成の核として機能していることが明らかとなった49).また,タバコ細胞においても,γチューブリン複合体に依存した新規微小管の伸長が誘導されること,フラグモプラスト周縁部でのこの新規微小管の伸長がフラグモプラストの拡大成長の推進力となっていることが報告された50).フラグモプラスト微小管の挙動を高解像度で解析した結果,フラグモプラスト微小管が,安定な微小管束と,動的で不安定な微小管により構成されていることが明らかとなった.γチューブリン複合体は,フラグモプラストの外縁に位置する安定な微小管束に結合しており,その複合体より新規微小管が伸長する.この新規に形成された動的微小管はフラグモプラストの外縁で架橋し,安定な微小管束になり,γチューブリン複合体の新たな結合場所を提供する.このようなフラグモプラスト外縁部の微小管束上に結合するγチューブリン複合体から繰り返し形成される新規微小管の形成が,フラグモプラストの拡大および,細胞質分裂の進行の原動力となっていることが示唆されている50).このようなフラグモプラスト微小管のダイナミクスの少なくとも一部は,先に説明したMAPキナーゼカスケードの下流で制御されているのであろう.
1)細胞板の起源
フラグモプラストの機能は,細胞板の材料を含む膜小胞をその赤道面に集積させること(細胞板の形成)および,その拡大に依存して細胞板を成長させること(細胞板の拡大成長)であると考えられている.つまり,細胞板の材料となる(膜成分と多糖類合成酵素などを含む)膜小胞の輸送は,フラグモプラスト微小管依存的であると推測されている.このことは,微小管重合阻害剤で処理すると,膜小胞が細胞板形成部位に集積せず細胞板が形成されない51),そして,フラグモプラスト微小管に膜小胞が付随している電子顕微鏡像が得られている14, 52, 53)という実験事実により支持されている.このような膜小胞の輸送にはキネシンの関与が指摘されている.しかし,多くのフラグモプラスト局在のキネシンが報告されているにも関わらず54),いまだ細胞板に膜小胞を輸送するキネシンは同定されていない.
電子顕微鏡による観察から,細胞板はゴルジもしくはトランスゴルジネットワーク(trans-Golgi network:TGN)由来の分泌小胞によって形成されると考えられてきた.しかし小胞体とゴルジ体間の輸送や細胞膜からのエンドサイトーシス経路を阻害する実験から,分泌小胞のみならずエンドサイトーシスも細胞板形成に重要であることが明らかとなっている55).エンドサイトーシスにより取り込まれた膜小胞は,初期エンドソーム(early endosome:EE)と呼ばれるコンパートメントに取り込まれる.最近の研究によりシロイヌナズナではTGNとEEは同一と考えられるようになったため,TGN/EEとも呼ばれるが,分泌とエンドサイトーシスの二つの小胞輸送システムは,植物ではいずれもTGN/EEを経由することが示されている56, 57).つまり,細胞板形成には,分泌小胞として新規に合成されるタンパク質とエンドサイトーシスにより輸送される成分がTGN/EEを経由して輸送される必要があると考えられている(図2B).
TGNから輸送される細胞板小胞の形成を制御する因子としてシロイヌナズナにおいてARF-GEF BIG1~4が同定された58).細胞内の単膜系オルガネラ間の輸送は,供与オルガネラからの輸送中間体(輸送小胞)の形成に始まり,標的オルガネラに運ばれた後に,繋留の過程を経て膜融合が起こることにより完了する.供与オルガネラからの輸送小胞の形成には,被覆複合体と呼ばれる分子群が機能しているが,BIG1~4タンパク質は,形成されたクラスリン被覆小胞の細胞板形成部位への輸送を仲介している.BIG1~4の機能阻害や,クラスリン被覆小胞の構成因子であるアダプタータンパク質複合体(AP1複合体)のサブユニットの除去により,分泌経路およびエンドサイトーシス経路の両方が阻害され,細胞板形成に異常が生じる58–60).これらの結果は,TGNおよび,分泌経路とエンドサイトーシス経路のいずれもが細胞板形成に寄与していることを示している.
2)細胞板形成における膜小胞の融合
被覆複合体の働きにより出芽した輸送小胞は,分裂面に輸送され,繋留因子(tethering protein)と呼ばれる分子群によって細胞板上に繋留される.Rab-GTPaseは,分子スイッチとしてこの繋留過程を制御する.繋留タンパク質複合体として同定されたシロイヌナズナのTRAPPII-TRAPPI-TRS120-TRS130は分裂面に局在し,それらの変異体は細胞質分裂異常を示す61–63).また,同じく繋留因子であるエクソシスト複合体(exocyst complex)も細胞板形成時の膜融合に関わっていることが知られている64).エクソシスト複合体に含まれるSEC6やEXO70A1は,細胞質分裂時に細胞板上に局在していることが報告されており,その変異体では細胞板の集合が異常となる65).上記した繋留タンパク質複合体の構成因子TRAPPIIの変異は,エクソシスト複合体の局在に影響を及ぼすことが報告されているので,両繋留因子は互いに関係しあっているのかもしれない.また,エクソシスト複合体の構成因子であるSEC6がKAULE(後述)と相互作用することも報告されているので66),繋留過程とその後のtrans-SNARE複合体形成の過程は物理的にリンクしていると考えられるが,これについてはまだよくわかっていない.
細胞質分裂に関与していることが明らかになっているRab-GTPaseは,A1~A3のサブグループに属するRab-GTPaseである.RABA2aとRABA2dはTGNと細胞板の周縁部に局在し,ドミナントネガティブの過剰発現では多核化した細胞を持つ細胞質分裂異常が観察される67).またRABA1cはTGNにおいてTRAPPII複合体のサブユニットであるTRS130と共局在することが示されている.さらにtrs130変異体では,RABA1cの局在が異常になることから,TRAPPII複合体がRAB1cの局在を規定しているのかもしれない65).
輸送小胞が細胞板に繋留されると,分裂面において膜小胞上のSNAREタンパク質と標的膜上のSNAREタンパク質が相互作用してtrans-SNARE複合体が形成され,これらの間で膜融合が起きる.SNAREタンパク質はQa-SNARE, Qb-SNARE, Qc-SNAREおよび,R-SNAREの四つに分類されるが,各SNAREタンパク質は共通のSNAREドメインを介して,他のSNAREタンパク質と相互作用し,さまざまな動的過程で特異的な複合体を形成して機能している.植物の細胞板形成時に特異的に合成され,重要な役割を果たしていることが知られているQa-SNARE KNOLLE/SYP111は,SNAP33(Qb-SNARE)-VAMP721/722(R-SNARE)および,NPSN11(Qb-SNARE)-SYP71(QcSNARE)-VAMP721/722の二つの複合体を形成することが明らかとなっているが,変異体の解析から,この二つの複合体は,いずれも細胞質分裂に寄与していることがわかった68, 69).また,細胞質分裂異常変異体として単離されたKAULEは,Qa-SNAREと結合するSec1/Munc18タンパク質の一つであり,SNAREタンパク質KNOLLEと結合し構造変化を誘導することにより,trans-SNARE形成を促進することが報告された70).このように,細胞板形成における膜融合の分子機構に関しては部分的には明らかになってきたが,細胞板小胞がどのように輸送されているのかはいまだブラックボックスである.フラグモプラストに沿った膜小胞輸送のメカニズムを明らかにすることが今後の課題である.
植物の細胞質分裂は細胞質分装置(細胞骨格)の動態制御と,細胞膜と細胞壁からなる細胞板の構築が共役して起こる必要がある.ここまで述べてきたように,個々の分子機構の一部は明らかになりつつあり,「はじめに」で述べたように,細胞板の拡大成長と共役したフラグモプラスト微小管の拡大成長との連携を示唆するデータが得られている.しかし,それぞれの過程を協調させる仕組みの解明はこれからである.最近,我々は,フラグモプラストの拡大成長を制御するMAPキナーゼカスケードの活性化因子であるシロイヌナズナのNACK1キネシンがフラグモプラストの赤道面(フラグモプラスト微小管のプラス端)に局在するためには,モータードメインだけでなく,C末端領域が重要であることを報告している71).このことは,NACK1がモーターキネシンとして機能している可能性と,C末端領域にそのモーター活性を制御する領域が存在することを示唆している.先にも述べたとおり,今のところ細胞板形成に関わる小胞を輸送するキネシンは同定されておらず,NACK1を含むフラグモプラスト微小管に局在するキネシンの詳細な分子機能の解明が待たれる.また最近,先に述べたSNAREタンパク質KNOLLEに相互作用するKEULEの変異体において,細胞板形成のみならずフラグモプラストの構造に異常がみられることが示され,このタンパク質が細胞板の形成と微小管ダイナミクスを協調させる因子として機能する可能性が提案されている72).今後,植物の細胞質分裂の分子機構の全体像を理解するためには,細胞板形成とフラグモプラストの拡大成長における共役的な制御機構や,細胞板形成において細胞膜と細胞壁の協調的形成を制御している分子機構の解明が重要であろう.
引用文献References
1) Yasuhara, H. & Shibaoka, H. (2000) Plant Cell Physiol., 41, 300–310.
2) Hülskamp, M., Parekh, N.S., Grini, P., Schneitz, K., Zimmermann, I., Lolle, S.J., & Pruitt, R.E. (1997) Dev. Biol., 187, 114–124.
3) Nishihama, R., Banno, H., Kawahara, E., Irie, K., & Machida, Y. (1997) Plant J., 12, 39–48.
4) Spielman, M., Preuss, D., Li, F.L., Browne, W.E., Scott, R.J., & Dickinson, H.G. (1997) Development, 124, 2645–2657.
5) Nishihama, R., Ishikawa, M., Araki, S., Soyano, T., Asada, T., & Machida, Y. (2001) Genes Dev., 15, 352–363.
6) Nishihama, R., Soyano, T., Ishikawa, M., Araki, S., Tanaka, H., Asada, T., Irie, K., Ito, M., Terada, M., Banno, H., Yamazaki, Y., & Machida, Y. (2002) Cell, 109, 87–99.
7) Strompen, G., El Kasmi, F., Richter, S., Lukowitz, W., Assaad, F.F., Jürgens, G., & Mayer, U. (2002) Curr. Biol., 12, 153–158.
8) Soyano, T., Nishihama, R., Morikiyo, K., Ishikawa, M., & Machida, Y. (2003) Genes Dev., 17, 1055–1067.
9) Yang, C.Y., Spielman, M., Coles, J.P., Li, Y., Ghelani, S., Bourdon, V., Brown, R.C., Lemmon, B.E., Scott, R.J., & Dickinson, H.G. (2003) Plant J., 34, 229–240.
10) Tanaka, H., Ishikawa, M., Kitamura, S., Takahashi, Y., Soyano, T., Machida, C., & Machida, Y. (2004) Genes Cells, 9, 1199–1211.
11) Ma, H.T. & Poon, R.Y. (2011) Biochem. J., 435, 17–31.
12) MAPK Group. (2002) Trends Plant Sci., 7, 301–308.
13) Hamel, L.P., Nicole, M.C., Sritubtim, S., Morency, M.J., Ellis, M., Ehlting, J., Beaudoin, N., Barbazuk, B., Klessig, D., Lee, J., Martin, G., Mundy, J., Ohashi, Y., Scheel, D., Sheen, J., Xing, T., Zhang, S., Seguin, A., & Ellis, B.E. (2006) Trends Plant Sci., 11, 192–198.
14) Yasuhara, H., Sonobe, S., & Shibaoka, H. (1993) Plant Cell Physiol., 34, 21–29.
15) Krysan, P.J., Jester, P.J., Gottwald, J.R., & Sussman, M.R. (2002) Plant Cell, 14, 1109–1120.
16) Sasabe, M., Boudolf, V., De Veylder, L., Inzé, D., Genschik, P., & Machida, Y. (2011) Proc. Natl. Acad. Sci. USA, 108, 17844–17849.
17) Takahashi, Y., Soyano, T., Kosetsu, K., Sasabe, M., & Machida, Y. (2010) Plant Cell Physiol., 51, 1766–1776.
18) Sazuka, T., Aichi, I., Kawai, T., Matsuo, N., Kitano, H., & Matsuoka, M. (2005) Plant Cell Physiol., 46, 1934–1943.
19) Naito, H. & Goshima, G. (2015) Cell Struct. Funct., 40, 31–41.
20) Calderini, O., Bögre, L., Vicente, O., Binarova, P., Heberle-Bors, E., & Wilson, C. (1998) J. Cell Sci., 111, 3091–3100.
21) Kosetsu, K., Matsunaga, S., Nakagami, H., Colcombet, J., Sasabe, M., Soyano, T., Takahashi, Y., Hirt, H., & Machida, Y. (2010) Plant Cell, 22, 3778–3790.
22) Beck, M., Komis, G., Müller, J., Menzel, D., & Samaj, J. (2010) Plant Cell, 22, 755–771.
23) Pellman, D., Bagget, M., Tu, Y.H., Fink, G.R., & Tu, H. (1995) J. Cell Biol., 130, 1373–1385.
24) Jiang, W., Jimenez, G., Wells, N.J., Hope, T.J., Wahl, G.M., Hunter, T., & Fukunaga, R. (1998) Mol. Cell, 2, 877–885.
25) Schuyler, S.C., Liu, J.Y., & Pellman, D. (2003) J. Cell Biol., 160, 517–528.
26) Müller, S., Smertenko, A., Wagner, V., Heinrich, M., Hussey, P.J., & Hauser, M.T. (2004) Curr. Biol., 14, 412–417.
27) Verbrugghe, K.J. & White, J.G. (2004) Curr. Biol., 14, 1755–1760.
28) Vernì, F., Somma, M.P., Gunsalus, K.C., Bonaccorsi, S., Belloni, G., Goldberg, M.L., & Gatti, M. (2004) Curr. Biol., 14, 1569–1575.
29) Mollinari, C., Kleman, J.P., Jiang, W., Schoehn, G., Hunter, T., & Margolis, R.L. (2002) J. Cell Biol., 157, 1175–1186.
30) Van Damme, D., Van Poucke, K., Boutant, E., Ritzenthaler, C., Inzé, D., & Geelen, D. (2004) Plant Physiol., 136, 3956–3967.
31) Wicker-Planquart, C., Stoppin-Mellet, V., Blanchoin, L., & Vantard, M. (2004) Plant J., 39, 126–134.
32) Mao, T., Jin, L., Li, H., Liu, B., & Yuan, M. (2005) Plant Physiol., 138, 654–662.
33) Sasabe, M., Soyano, T., Takahashi, Y., Sonobe, S., Igarashi, H., Itoh, T.J., Hidaka, M., & Machida, Y. (2006) Genes Dev., 20, 1004–1014.
34) Caillaud, M.C., Lecomte, P., Jammes, F., Quentin, M., Pagnotta, S., Andrio, E., de Almeida Engler, J., Marfaing, N., Gounon, P., Abad, P., & Favery, B. (2008) Plant Cell, 20, 423–437.
35) Smertenko, A.P., Chang, H.Y., Sonobe, S., Fenyk, S.I., Weingartner, M., Bögre, L., & Hussey, P.J. (2006) J. Cell Sci., 119, 3227–3237.
36) Sasabe, M., Kosetsu, K., Hidaka, M., Murase, A., & Machida, Y. (2011) Plant Signal. Behav., 26, 743–747.
37) Ito, M., Iwase, M., Kodama, H., Lavisse, P., Komamine, A., Nishihama, R., Machida, Y., & Watanabe, A. (1998) Plant Cell, 10, 331–341.
38) Ito, M. (2000) Plant Mol. Biol., 43, 677–690.
39) Ito, M., Araki, S., Matsunaga, S., Itoh, T., Nishihama, R., Machida, Y., Doonan, J.H., & Watanabe, A. (2001) Plant Cell, 13, 1891–1905.
40) Ito, M. (2005) J. Plant Res., 118, 61–69.
41) Haga, N., Kobayashi, K., Suzuki, T., Maeo, K., Kubo, M., Ohtani, M., Mitsuda, N., Demura, T., Nakamura, K., Jürgens, G., & Ito, M. (2011) Plant Physiol., 157, 706–717.
42) 伊藤正樹,笹部美知子,町田泰則(2016)領域融合レビュー,5, e005.
43) Araki, S., Ito, M., Soyano, T., Nishihama, R., & Machida, Y. (2004) J. Biol. Chem., 279, 32979–32988.
44) Weingartner, M., Criqui, M.C., Mészáros, T., Binarova, P., Schmit, A.C., Helfer, A., Derevier, A., Erhardt, M., Bögre, L., & Genschik, P. (2004) Plant Cell, 16, 643–657.
45) Hepler, P.K. & Wolniak, S.M. (1984) Int. Rev. Cytol., 90, 169–238.
46) Baskin, T.I. & Cande, W.Z. (1990) Annu. Rev. Plant Physiol. Plant Mol. Biol., 41, 277–315.
47) Zhang, D., Wadsworth, P., & Hepler, P.K. (1993) Cell Motil. Cytosk., 24, 151–155.
48) Goshima, G., Mayer, M., Zhang, N., Stuurman, N., & Vale, R.D. (2008) J. Cell Biol., 181, 421–429.
49) Nakaoka, Y., Miki, T., Fujioka, R., Uehara, R., Tomioka, A., Obuse, C., Kubo, M., Hiwatashi, Y., & Goshima, G. (2012) Plant Cell, 24, 1478–1493.
50) Murata, T., Sano, T., Sasabe, M., Nonaka, S., Higashiyama, T., Hasezawa, S., Machida, Y., & Hasebe, M. (2013) Nat. Commun., 4, 1967.
51) Palevitz, B.A. & Hepler, P.K. (1974b) Chromosoma, 46, 327–341.
52) Kakimoto, T. & Shibaoka, H. (1988) Protoplasma, 2(Suppl 2), 95–103.
53) Segui-Simarro, J.M., Austin, J.R. II, White, E.A., & Staehelin, L.A. (2004) Plant Cell, 16, 836–856.
54) Miki, T., Naito, H., Nishina, M., & Goshima, G. (2014) Proc. Natl. Acad. Sci. USA, 111, E1053–E1061.
55) Reichardt, I., Stierhof, Y.D., Mayer, U., Richter, S., Schwarz, H., Schumacher, K., & Jürgens, G. (2007) Curr. Biol., 17, 2047–2053.
56) Dettmer, J., Hong-Hermesdorf, A., Stierhof, Y.D., & Schumacher, K. (2006) Plant Cell, 18, 715–730.
57) Viotti, C., Bubeck, J., Stierhof, Y.D., Krebs, M., Langhans, M., van den Berg, W., van Dongen, W., Richter, S., Geldner, N., Takano, J., Jürgens, G., de Vries, S.C., Robinson, D.G., & Schumacher, K. (2010) Plant Cell, 22, 1344–1357.
58) Richter, S., Kientz, M., Brumm, S., Nielsen, M.E., Park, M., Gavidia, R., Krause, C., Voss, U., Beckmann, H., Mayer, U., Stierhof, Y.D., & Jürgens, G. (2014) eLife, 3, e02131.
59) Park, M., Song, K., Reichardt, I., Kim, H., Mayer, U., Stierhof, Y.D., Hwang, I., & Jürgens, G. (2013) Natl. Acad. Sci. USA, 110, 10318–10323.
60) Teh, O.K., Shimono, Y., Shirakawa, M., Fukao, Y., Tamura, K., Shimada, T., & Hara-Nishimura, I. (2013) Plant Cell Physiol., 54, 838–847.
61) Jaber, E., Thiele, K., Kindzierski, V., Loderer, C., Rybak, K., Jürgens, G., Mayer, U., Söllner, R., Wanner, G., & Assaad, F.F. (2010) New Phytol., 187, 751–763.
62) Thellmann, M., Rybak, K., Thiele, K., Wanner, G., & Assaad, F.F. (2010) Plant Physiol., 154, 720–732.
63) Qi, X., Kaneda, M., Chen, J., Geitmann, A., & Zheng, H. (2011) Plant J., 68, 234–248.
64) Fendrych, M., Synek, L., Pecenková, T., Toupalová, H., Cole, R., Drdová, E., Nebesárová, J., Sedinová, M., Hála, M., Fowler, J.E., & Zársky, V. (2010) Plant Cell, 22, 3053–3065.
65) Rybak, K., Steiner, A., Synek, L., Klaeger, S., Kulich, I., Facher, E., Wanner, G., Kuster, B., Zarsky, V., Persson, S., & Assaad, F.F. (2014) Dev. Cell, 29, 607–620.
66) Wu, J., Tan, X., Wu, C., Cao, K., Li, Y., & Bao, Y. (2013) Mol. Plant, 6, 1863–1876.
67) Chow, C.M., Neto, H., Foucart, C., & Moore, I. (2008) Plant Cell, 20, 101–123.
68) El Kasmi, F., Krause, C., Hiller, U., Stierhof, Y.D., Mayer, U., Conner, L., Kong, L., Reichardt, I., Sanderfoot, A.A., & Jürgens, G. (2013) Mol. Biol. Cell, 24, 1593–1601.
69) Kwon, C., Neu, C., Pajonk, S., Yun, H.S., Lipka, U., Humphry, M., Bau, S., Straus, M., Kwaaitaal, M., Rampelt, H., El Kasmi, F., Jürgens, G., Parker, J., Panstruga, R., Lipka, V., & Schulze-Lefert, P. (2008) Nature, 451, 835–840.
70) Park, M., Touihri, S., Müller, I., Mayer, U., & Jürgens, G. (2012) Dev. Cell, 22, 989–1000.
71) Sasabe, M., Ishibashi, N., Haruta, T., Minami, A., Kurihara, D., Higashiyama, T., Nishihama, R., Ito, M., & Machida, Y. (2015) J. Plant Res., 128, 327–336.
72) Steiner, A., Müller, L., Rybak, K., Vodermaier, V., Facher, E., Thellmann, M., Ravikumar, R., Wanner, G., Hauser, M.T., & Assaad, F.F. (2016) Mol. Plant, 9, 528–540.
著者紹介Author Profile
 笹部 美知子(ささべ みちこ)
笹部 美知子(ささべ みちこ)弘前大学農学生命科学部准教授.博士(学術).
略歴岡山県岡山市出身.1997年岡山大学農学部卒業.2001年岡山大学大学院自然科学研究科博士後期課程修了.日本学術振興会特別研究員,名古屋大学大学院理学研究科研究員,中部大学植物センター研究員を経て07年名古屋大学大学院理学研究科特任助教.12年より現職.
研究テーマと抱負植物の細胞分裂を制御する分子メカニズムの研究.細胞分裂と細胞の運命決定のメカニズムに少しでも近づきたいと思いながら研究をしています.
趣味ドライブ,音楽鑑賞.
 町田 泰則(まちだ やすのり)
町田 泰則(まちだ やすのり)名古屋大学大学院理学研究科研究員(名誉教授).理学博士.
略歴1948年群馬県に生る.72年千葉大学理学部卒業.78年名古屋大学大学院理学研究科博士課程満了,2012年名古屋大学大学院教授を定年退職し,特任教授をへて16年より現職.
研究テーマと抱負細胞の分裂と分化の仕組みを分子レベルから研究する.最近これらの仕組みと核小体(およびその周辺領域)の構築と機能とが連関していると考えるようになり,新規な方法論の必要性を感じている.
ウェブサイトhttp://www.bio.nagoya-u.ac.jp/~yas/dmcb/dmcb.html
趣味水泳.