我々の体は常に外来微生物の侵入による脅威にさらされており,これらから身を守っているのが免疫システムである.この免疫システムの中でも最前線に位置するのが自然免疫であり,外敵の侵入をすばやく感知し,初期の免疫応答やそれに引き続く獲得免疫を活性化させる役割を果たす.パターン認識受容体と呼ばれる免疫受容体が微生物の構成成分の分子パターンを認識し活性化することで,下流の分子にシグナルを伝達する1).代表的なパターン認識受容体としてToll様受容体(Toll-like receptor:TLR),NOD様受容体,RIG-I様受容体などが知られている2).筆者らのグループは長年TLR受容体の構造生物学的研究に取り組んでおり,TLR4-MD-2, TLR8, TLR9に関してそのリガンドとの複合体の構造解析に成功した.本稿では,これらの結果に関して紹介したい.
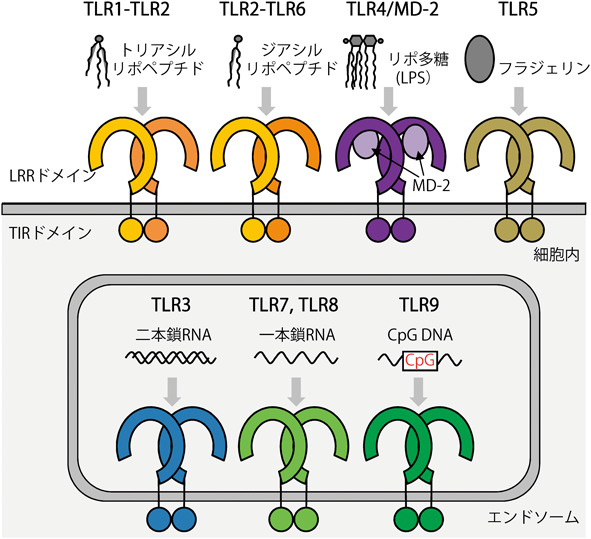
TLRはヒトでは10種類(TLR1~TLR10),マウスでは12種類(TLR1~TLR9とTLR11~TLR13)が同定されており,それぞれ異なる病原体由来の分子パターンをリガンドとして認識して活性化する(図1).TLR1, TLR2, TLR6はリポペプチド3–6),TLR4は共受容体MD-2とともにリポ多糖(LPS)7–10),TLR5は鞭毛の構成タンパク質であるフラジェリン11, 12),TLR3は二本鎖RNA13),TLR7とTLR8は一本鎖RNA14–16),TLR9は非メチル化CpGモチーフを有するDNA17)を認識する.TLRは一回膜貫通型の受容体で細胞外のロイシンリッチリピート(LRR)ドメイン,膜貫通部位,細胞内のTIRドメインから構成されている18, 19).LRRは20~30残基程度の繰り返し配列が連なった構造モチーフであり,一般的には一つのLRR単位は1組のβ鎖とαへリックスまたはループ構造からなり,全体として凹面がβシートにより構成される馬蹄形構造を形成する.TLRのLRRドメインは20~26のLRR単位から構成されており,リガンド結合を担うとされている.一般的には,リガンド非結合状態のTLRは単量体として存在し,リガンドが結合することで二量体化して活性化すると考えられている20)(図1).
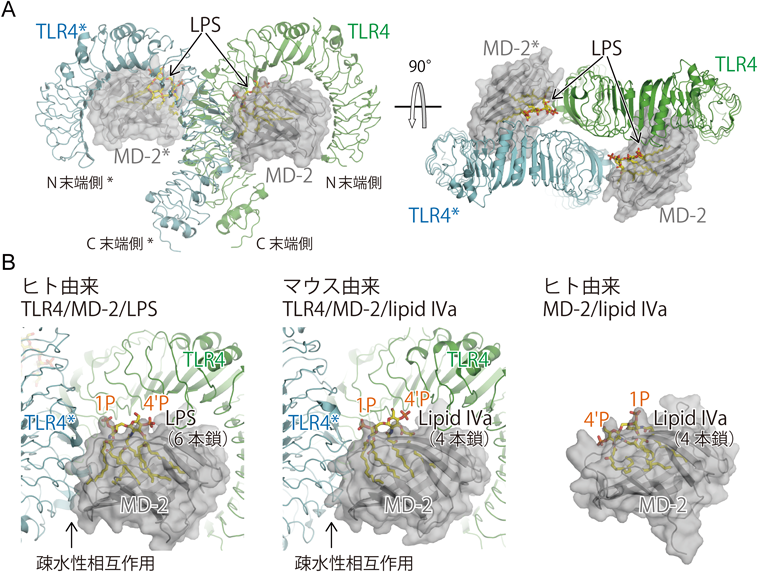
グラム陰性細菌の外膜の構成成分のLPSはエンドトキシンとも呼ばれ,非常に強力な免疫応答を引き起こす(図1).それゆえに過度のLPS応答は,エンドトキシンショックや敗血症などの全身性の炎症反応を引き起こし,しばしば多臓器不全などの生命を脅かす重篤な症状となる.そのため,その根本的な治療法の開発が待望されている.LPSを認識する受容体としてまずTLR4が同定された8–10).その後,TLR4は可溶性の160残基程度のタンパク質MD-2と複合体を形成してLPSを認識することが示された7, 21)(図2).筆者らはまずMD-2に着目しその構造決定を進め,ヒトMD-2に関してリガンド非添加型およびLPSのアンタゴニストであるlipid IVaとの複合体の2種類の構造を明らかにした22)(図2B右).MD-2は二つのβシートからなる免疫グロブリンフォールドの構造をとっており,シートの間には疎水性のポケットが存在していた.興味深いことに,リガンド非添加の構造においてもポケットの内部に脂質由来と思われる電子密度が観察された.lipid IVaが有する4本の脂肪酸鎖はすべてMD-2のポケット内部にはまり込む形で結合していた(図2B右).この構造から,免疫活性を有する大腸菌型のLPSは6本の脂肪酸鎖を有しているが,その場合も同様に脂肪酸鎖がMD-2のポケットに結合するであろうこと,つまり,TLR4/MD-2複合体においてMD-2がLPSの直接の結合部位であることが強く示唆された.その後,TLR4/MD-2複合体の結晶構造23)およびヒトTLR4/MD-2/LPS三者複合体の結晶構造24)が韓国のグループより報告された(図2A).TLR4/MD-2複合体は1:1の,TLR4/MD-2/LPS複合体は2:2:2の複合体を形成していた.なお本稿では,二量体を形成するTLRの2分子の一方をTLR,もう一方をTLR*と記述する.MD-2はTLR4の凹面側に結合し馬蹄形構造が作る面から半分突き出す形で結合していた(図2A右).LPSはMD-2の疎水的ポケットに脂肪酸鎖をはめ込む形で結合していた(図2A, 2B左).アンタゴニストであるlipid IVaでは4本の脂肪酸鎖すべてがMD-2の疎水的なポケットに収納されていた(図2B右)のに対して,6本鎖のLPSでは5本の脂肪酸鎖がポケットに収納され残りの1本の脂肪酸鎖がポケットの入り口に沿う形で表面に露出していた(図2B左).そして露出した疎水性部位がもう1分子のTLR4*の凸面に存在する疎水性部分と相互作用することで,全体として2:2:2複合体を形成していた.複合体中の2分子のTLR4は互いのN末端側を外側にC末端側を中央に向けたM字形の配置をしていた(図2A左).この配置により2分子のTLR4の細胞外ドメインのC末端どうしは約20 Åにまで接近していた.これにより,細胞内のTIRドメインが互いに相互作用し,他のアダプター分子のTIRドメインとのTIRドメイン間の相互作用の足場として働くことでシグナルが伝達されることが予想される.筆者らのグループはさらにマウスTLR4/MD-2と4本鎖のlipid IVaとの複合体の結晶構造解析に成功した25)(図2B中).lipid IVaはヒトTLR4/MD-2に対してはアンタゴニストとして作用するが,マウスTLR4/MD-2に対しては弱いアゴニストとして作用する26, 27).構造解析の結果,マウスTLR4/MD-2/lipid IVa複合体はLPSとの複合体と同様の配置の2:2:2複合体を形成していた.この際,lipid IVaはヒトMD-2に結合した場合とはまったく異なる配置でマウスMD-2のポケットに結合していた(図2B中,右).リガンド全体が約180度回転した形で結合しており,4本の脂肪酸鎖のMD-2のポケット内での配置もまったく異なっていた.ヒトMD-2に結合した場合よりも,脂肪酸鎖がポケットに浅く結合しており,ポケットの浅い側に位置する脂肪酸鎖が一部ポケット表面に露出していた(図2B中).このため,lipid IVaの場合でももう1分子のTLR4*に対する疎水的表面を提供することで2:2:2の複合体形成が可能になると考えられる.ただしこの脂肪酸鎖の配置では,ポケットの内部が一部満たされておらず,6本鎖のLPSに比べて二量体構造が不安定で,活性が弱いことが考えられる.実際,6本鎖LPSとの三者複合体はゲルろ過クロマトグラフィーなどで検出できるが,4本鎖lipid IVaとの三者複合体は溶液中では観察されず結晶中というきわめて高濃度でのみ観察された.これまでlipid IVaに対する種特異的な応答に関して,主に変異体を用いてさまざまな解析がなされてきた28–34).それらの結果から,lipid IVaの種特異性に重要な残基は,MD-2だけでなくTLR4にも存在することが明らかになっている.構造解析の結果と合わせて考えると,MD-2とTLR4*の相互作用面周辺のTLR4の表面電荷の違い,MD-2の疎水性の違い,MD-2のポケット入り口付近の電荷の違い,MD-2のポケットの微妙な形状の違い,などが複合的に影響した結果,lipid IVaに対する特異性が生み出されていると考えられる.
TLR7, TLR8, TLR9はTLR7サブファミリーに属し,TLRの中でも高い相同性を有し,いずれも一本鎖核酸を認識する(図1).TLR7とTLR8は抗ウイルス作用を示すイミダゾキノリン系の低分子合成リガンドにより活性化されることがまず明らかになり35, 36),その後ウイルス由来のRNAにより活性化されることが明らかにされ14–16),一般的には一本鎖RNAを認識するTLRとされている.TLRのリガンドの中で最も分子量の小さい合成リガンドによって活性化されることから,TLR7とTLR8は治療薬ターゲットとして有望視されている.TLR7サブファミリーに属するTLRの細胞外ドメインは,TLRの中でも最も長い26のLRR単位から構成され,LRR14とLRR15の間に40残基程度の挿入ループ領域を有している19).以降この領域をZ-loopと呼ぶ.興味深いことにTLR7サブファミリーの機能制御にZ-loopが関与しているとの論文が次々と報告された37–41).それらによるとZ-loopが切断されることで活性化能を有する成熟型TLRとなり,切断にはリソソームに存在するカテプシンなどの種々のプロテアーゼが関与するとされている.このことは,TLR7サブファミリーが働く場であるリソソームに到達する前はZ-loopが未切断状態であり,自己の核酸に対して応答するのを防ぐ役割があるものと考えられる.
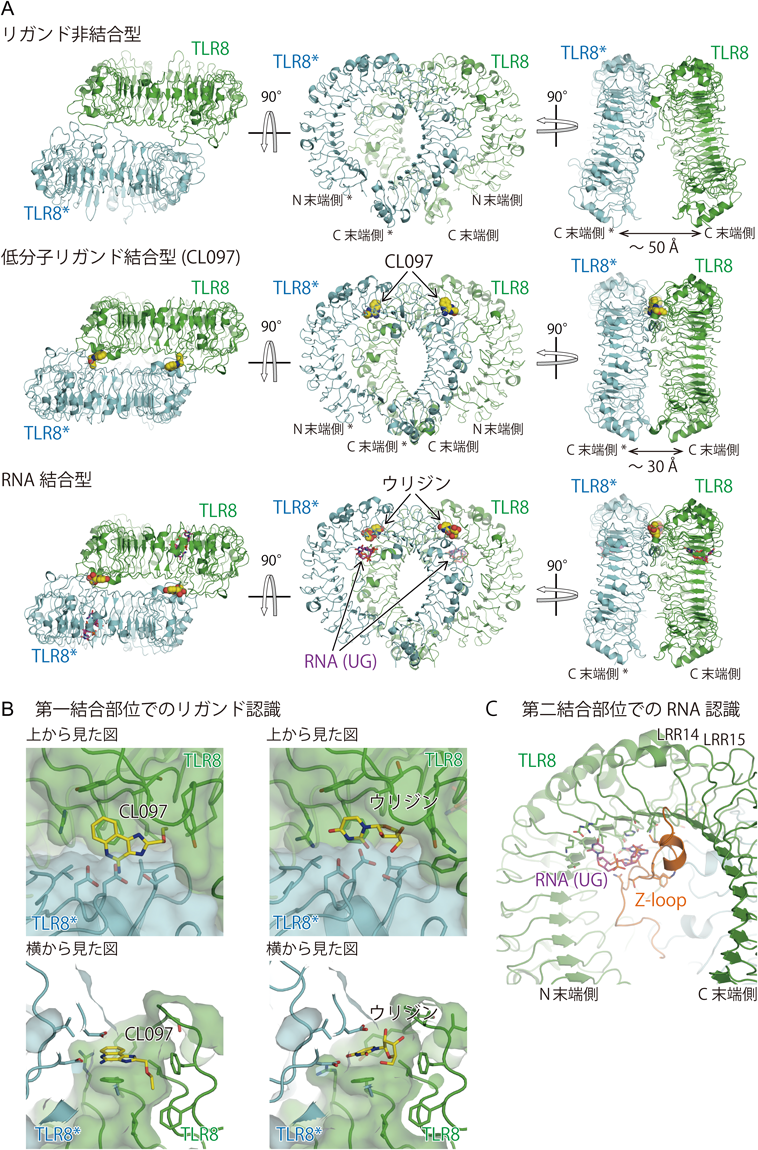
筆者らはまずTLR8単独と合成リガンドとの複合体の結晶構造解析を行った42)(図3A上,中).溶液状態での性状解析の結果,ヒトTLR8の細胞外ドメインは単独ですでに二量体を形成していることが明らかになった.さらに,合成リガンドを加えることで二量体の構造変化が起きることがX線小角散乱パターンから示唆されていた.実際,結晶中においてもリガンド非結合状態の二量体構造が観察された.また,合成リガンド(CL097, CL075, R848)結合状態でも二量体構造が観察された.両者の二量体構造を比較すると,いずれもTLR4/MD-2/LPS複合体と同様に2分子のTLR8のC末端どうしが向かい合う形の二量体構造であることは共通していたが,リガンド非結合型ではC末端どうしの距離が約50 Å離れていたのに対して,合成リガンド結合型ではC末端どうしの距離が約30 Åと接近していた(図3A上,中).このことはリガンド結合型の二量体構造が活性化型構造であることを示唆している.リガンド非結合型とリガンド結合型のTLR8の二量体構造を比較すると二量体を形成する相互作用は両者の間でまったく異なっていた.つまり,TLR8はリガンドが結合することで二量体構造を再構成して活性化型構造となることが明らかになった.リガンドはTLR8とTLR8*にはさまれる形で二量体の等価な2か所に結合しており,リガンドを介して活性化型のTLR8の二量体構造が安定化されていた(図3A中).
結晶化に用いたヒトTLR8の細胞外ドメインはショウジョウバエS2細胞で発現させたものであり,発現させたTLR8はすでにZ-loop部分で切断されSDS-PAGE上では2本のポリペプチド鎖として観察されている42).しかし,N末端側とC末端側の断片が精製過程で解離することはなく,両者は強固に相互作用していることが予想された.実際に,TLR8の二量体を構成するプロトマーの構造では,N末端側とC末端側の断片は互いに相互作用して一つのリング型構造を形成していた.LRR14とLRR15はZ-loopをはさんでいるが,一連のLRR構造を形成していた.また,TLR8のN末端とC末端は直接相互作用していた.さらに,切断されたZ-loopの後半部分(C末端断片に含まれる)はN末端断片のLRR構造の凹面と広範に疎水性の相互作用を形成していた.TLR8と合成リガンドとの複合体の構造解析の結果,リガンドはTLR8のN末端側断片とTLR8*のC末端側断片の間にはさまれる形で結合しており(図3A中),Z-loopで切断後の二つの断片の両方がリガンド結合に重要な役割を果たすことが構造的に実証された.
前述のようにTLR8は一本鎖RNAの受容体として一般に認識されている.しかし,合成リガンドとRNAはその大きさも化学的性質もまったく異なり,一本鎖RNAによってTLR8が活性化される機構を合成リガンドとの複合体構造から予測するのは困難であった.そこで筆者らは,TLR8と一本鎖RNAとの複合体の結晶構造解析を行い,その結果,TLR8は一本鎖RNAとウリジンを同時に認識して活性化することが明らかになった.
TLR8と20merの一本鎖RNAとの共結晶化を行った結果,合成リガンド結合型と同様の活性化型二量体構造が観察された43)(図3A下).予想外にも,結晶構造中に20merのRNAに相当する長さの電子密度は観察されなかった.その代わりにウリジンと短いRNA(UGジヌクレオチド)に相当する電子密度が2か所の結合部位に観察された(図3A下,3B, 3C).ウリジンは合成リガンドが結合していた場所に結合していた(第一結合部位と呼ぶ,図3B右).RNA(UG)は,TLR8のZ-loop部分とリング型構造の内側の凹面にはさまれる形で結合していた(第二結合部位と呼ぶ,図3C).これらの2種類のリガンドが,20merのRNAの一部なのか,RNAが分解して生じた断片なのかを明らかにするために,結晶中に含まれる核酸成分の高速液体クロマトグラフ質量分析(LC-MS)を行った.その結果,20merのRNAはまったく検出されず,ウリジンとUG, UUGなどの短鎖RNAが検出された.つまり,20merのRNAそのものがTLR8に結合して活性化型二量体を誘導しているのではなく,RNAの分解産物がTLR8の2か所の結合部位に作用して活性化型二量体を誘導していることが明らかになった.第一結合部位は二量体界面に存在し活性化型二量体構造の安定化に直接寄与するのに対して,第二結合部位は二量体界面からは離れた位置に存在しており活性化型二量体構造形成には直接は寄与していないことが予想された(図3A下).
TLR8と一本鎖RNAの構造解析の結果より,筆者らは二つの仮説を考えた.一つ目は,TLR8はウリジンを認識する受容体なのではないかということ.二つ目は,TLR8がウリジンを認識する際にRNAは補助的に作用するのではないかということである.これらの仮説は,等温滴定カロリメトリー(ITC)およびHEK293T細胞を用いたNF-κBリポーター遺伝子アッセイにより検証された.ITCの結果,種々のモノヌクレオシドの中で唯一ウリジンの場合だけTLR8との結合熱が観測された.また,5′位または3′位にリン酸基が付加したUMPではまったく結合熱が観測されなかった.これは,ウリジンが結合したTLR8の構造中にさらに余分なリン酸基を収納する空間が存在しないことと対応している(図3B右).TLR8とウリジンとの間の解離定数は約50 μMであり,合成リガンドの一つであるR848との解離定数約0.2 μMと比較するとそれほど強いものではなかった.つまり,TLR8はモノヌクレオシドの中でウリジンに特異的に結合は示すものの,合成リガンドに比べると圧倒的に結合が弱く,それ自身ではTLR8を活性化するのに不十分であることが予想された.そこで,構造解析の結果から,第二結合部位に短鎖RNAが結合していたことを考慮して,TLR8と構造解析に使用した一本鎖RNAを混合した溶液にウリジンを滴定したところ,ウリジンとの間の解離定数は約1 μMと約50倍に結合が増強された.同様にHEK293T細胞を用いたNF-κBリポーター遺伝子アッセイでは,各種モノヌクレオシド単独および(単独では活性を示さない低濃度での)一本鎖RNAと共刺激を行ったところ,ウリジンと一本鎖RNAで共刺激した場合にだけ顕著にTLR8の活性化が確認された.これらの結果より,第二結合部位に一本鎖RNAが結合することで第一結合部位へのウリジンの結合が促進されるという活性化モデルが提唱された43).
合成リガンドとウリジンはいずれもTLR8の第一結合部位に結合するが,両者とTLR8の間の相互作用には共通点および相違点がみられた(図3B).合成リガンドはTLR8と三つの特徴的な相互作用,すなわちTLR8の芳香環を持つ残基とリガンドとの間のスタッキング相互作用,リガンドのアミジノ基とTLR8の保存されたアスパラギン酸残基の間の水素結合,さらにリガンドの疎水性の置換基とTLR8の二つのプロトマーによって形成された疎水性のポケットの間の相互作用を形成していた(図3B左).ウリジンの認識においても,スタッキング相互作用とアスパラギン酸残基との水素結合が形成されていたが,疎水性のポケットとの間の相互作用はみられなかった(図3B右).ウリジンとTLR8の相互作用の場合には,この疎水性相互作用が形成できないことが合成リガンドに比べて結合が弱い一因であろう.
4. CpGモチーフを有するDNAを認識するTLR9受容体
微生物が有するCpGモチーフ配列を有するDNA(CpG DNA)は免疫系を活性化することが知られていた44).その後,CpG DNAを認識して活性化する受容体がTLR9であることが見いだされた17)(図1).哺乳動物由来のCpG配列は高頻度でメチル化されており免疫活性化が低いのに対して,微生物由来のCpG配列はメチル化されていない.そのため,TLR9はメチル化CpG配列を見分けることで自己と非自己を識別していると考えられている.これまでに,主に培養細胞を用いて種々のDNA配列の免疫活性化能が調べられてきたが,CpG DNAがTLR9のどこにどのように作用するのか,なぜメチル化の有無で免疫活性化能が変化するのかは不明であった.TLR9を刺激するCpG DNAはワクチンのアジュバンドとして活用が期待されており,その機能をより効果的に制御するにはTLR9とCpG DNAとの相互作用を原子レベルで明らかにする必要がある.そこで,筆者らはTLR9のリガンド非結合型,アゴニスト(CpG DNA)結合型,さらにアンタゴニストDNA結合型の3種類の結晶構造を明らかにした45).
筆者らはさまざまな動物種由来のTLR9の発現をスクリーニングし,マウス,ウシ,ウマTLR9に関してリコンビナントタンパク質を得て,結晶構造解析を進めた46).なお,TLR8では発現させた時点ですでにZ-loopで切断されていたのに対して,TLR9は切断されていなかったため,V8プロテアーゼを作用させることでZ-loopで切断されたTLR9を得た.Z-loop未切断のTLR9および切断したTLR9を用いてゲルろ過クロマトグラフィーと超遠心分析により溶液状態の会合状態およびDNAとの相互作用を調べた.その結果,TLR9はリガンド非結合状態では単量体として存在しており,またTLR9のZ-loopの切断の有無でDNA結合能に違いはみられなかった.しかし,Z-loop未切断体ではDNAを結合させても単量体のままであるのに対して,Z-loop切断体ではCpG DNA依存的なTLR9の二量体化が確認された.つまり,TLR9のZ-loop切断はDNA結合には関係しないが,その後のTLR9の二量体形成に影響することが明らかになった.Z-loopがTLRの二量体形成を阻害することに関して,筆者らはZ-loopが切断されないように変異を入れることで未切断体のTLR8を調製しその性状解析と構造解析を行った46).TLR8においてもTLR9と同様にZ-loop未切断体ではRNAの結合能は有するが二量体形成はみられなかった.さらに,Z-loop未切断体のTLR8の構造解析を行い,未切断のZ-loopがTLR8の二量体界面側に位置することで二量体形成を阻害していることを明らかにした.
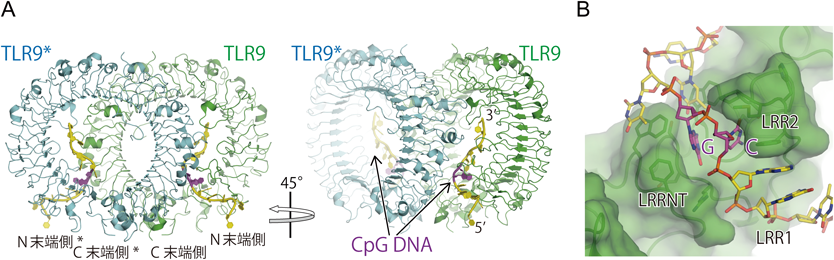
構造解析の結果,リガンド非結合状態のTLR9は単量体として存在し,その構造はTLR8の二量体のプロトマー構造とよく似ていた46).TLR9はZ-loopで切断されていたが,やはりN末端側とC末端側の断片は互いに相互作用して一つのリング型構造を形成していた.CpG DNAとして12merのDNAを用いてTLR9との複合体の結晶構造を明らかにした(図4A).CpG DNAとTLR9は2:2の複合体を形成し,その配置はTLR8の活性化型二量体構造と非常によく似ていた.しかし,CpG DNAの結合部位はまったく異なり,TLR9のN末端側の側面からリングの内側へと巻きつくような形で結合し,同時にTLR9*のC末端側断片の凸面と相互作用していた(図4A).つまりCpG DNAは2分子のTLR9にはさまれる形で結合し,活性化型二量体構造を安定化していた.CpGモチーフ部分はTLR9のN末端側(LRR-NT, LRR1, LRR2)に存在する溝にはまり込む形で結合していた(図4B).特にCpGモチーフのシトシンとグアニンの塩基部分はいずれもTLR9と密にファンデルワールス接触しており,また水素結合を形成していた.このCpGモチーフ部分の認識がTLR9との結合に重要であることを示すために,CG配列部分を別の配列(GC, TC, CA, UG,メチル化CG)に置換した配列を用いてITC実験を行った.予想どおり,CGのとき最も結合が強く,他の配列では結合が減弱した.また,CGをメチル化することで結合が減弱することも確認された.興味深いことに,TLR9のDNA結合はpH依存性を示した.酸性pH条件下では,TLR9とDNAは強固に結合するのに対して,中性以上のpH条件下では結合は著しく減弱した.TLR9とDNAとの結合には,中性付近にpKaを有するヒスチジン残基が多数関与しており,これらのヒスチジン残基のプロトン化が結合に重要であることが示唆される.同様のpH依存的な核酸の結合はTLR3においても報告されている47, 48).
筆者らはアゴニストDNA(CpG DNA)結合型のTLR9構造に加えて,アンタゴニストDNA結合型TLR9構造を明らかにした46).アンタゴニストDNAは分子内で水素結合を形成しステムループ構造をとりTLR9のリング型構造の内側に結合していた.アンタゴニストDNAの結合部位はアゴニストDNAと一部重なっており,アゴニストDNAよりも強固にTLR9に結合することで競合的にTLR9の活性化を阻害するものと考えられる.
TLRの機能が最初に明らかにされてからおよそ20年が経過した.TLRの機能発現を理解する上で構造生物学的研究の果たした役割は大きい.それまで漠然としていたTLRの活性化機構について,TLRとそのリガンドとの複合体の構造解明はその詳細なリガンド認識機構を明らかにしただけでなく,未知のリガンド候補物質との相互作用に関してもある程度の予測をすることを可能にした.また,TLR8とRNAとの複合体の構造解析でみられたように,構造解析して初めて真の意味でのリガンドが同定されることもある.これらの結果が,今後TLRの機能を制御する化合物の開発の一助となることが期待される.
謝辞Acknowledgments
本研究は東京大学大学院薬学系研究科蛋白構造生物学教室(清水敏之教授)において行われたものである.清水教授および共に研究を進めた研究室のメンバーに心より感謝いたします.また,本研究は東大医科研の三宅健介教授,柴田琢磨博士(細胞を用いたアッセイ),阪大の内山進博士(超遠心分析),首都大学東京の礒辺俊明教授(LC-MS)らの研究グループとの共同研究であり,放射光実験では,SPring-8およびPhoton Factoryのビームラインスタッフの方々に大変お世話になりました.心より感謝いたします.
引用文献References
1) Akira, S., Uematsu, S., & Takeuchi, O. (2006) Cell, 124, 783–801.
2) Kawai, T. & Akira, S. (2009) Int. Immunol., 21, 317–337.
3) Takeuchi, O., Sato, S., Horiuchi, T., Hoshino, K., Takeda, K., Dong, Z.Y., Modlin, R.L., & Akira, S. (2002) J. Immunol., 169, 10–14.
4) Alexopoulou, L., Thomas, V., Schnare, M., Lobet, Y., Anguita, J., Schoen, R.T., Medzhitov, R., Fikrig, E., & Flavell, R.A. (2002) Nat. Med., 8, 878–884.
5) Takeuchi, O., Kawai, T., Muhlradt, P.F., Morr, M., Radolf, J.D., Zychlinsky, A., Takeda, K., & Akira, S. (2001) Int. Immunol., 13, 933–940.
6) Ozinsky, A., Underhill, D.M., Fontenot, J.D., Hajjar, A.M., Smith, K.D., Wilson, C.B., Schroeder, L., & Aderem, A. (2000) Proc. Natl. Acad. Sci. USA, 97, 13766–13771.
7) Shimazu, R., Akashi, S., Ogata, H., Nagai, Y., Fukudome, K., Miyake, K., & Kimoto, M. (1999) J. Exp. Med., 189, 1777–1782.
8) Qureshi, S.T., Lariviere, L., Leveque, G., Clermont, S., Moore, K.J., Gros, P., & Malo, D. (1999) J. Exp. Med., 189, 615–625.
9) Hoshino, K., Takeuchi, O., Kawai, T., Sanjo, H., Ogawa, T., Takeda, Y., Takeda, K., & Akira, S. (1999) J. Immunol., 162, 3749–3752.
10) Poltorak, A., He, X.L., Smirnova, I., Liu, M.Y., Van Huffel, C., Du, X., Birdwell, D., Alejos, E., Silva, M., Galanos, C., Freudenberg, M., Ricciardi-Castagnoli, P., Layton, B., & Beutler, B. (1998) Science, 282, 2085–2088.
11) Uematsu, S., Jang, M.H., Chevrier, N., Guo, Z.J., Kumagai, Y., Yamamoto, M., Kato, H., Sougawa, N., Matsui, H., Kuwata, H., Hemmi, H., Coban, C., Kawai, T., Ishii, K.J., Takeuchi, O., Miyasaka, M., Takeda, K., & Akira, S. (2006) Nat. Immunol., 7, 868–874.
12) Hayashi, F., Smith, K.D., Ozinsky, A., Hawn, T.R., Yi, E.C., Goodlett, D.R., Eng, J.K., Akira, S., Underhill, D.M., & Aderem, A. (2001) Nature, 410, 1099–1103.
13) Alexopoulou, L., Holt, A.C., Medzhitov, R., & Flavell, R.A. (2001) Nature, 413, 732–738.
14) Lund, J.M., Alexopoulou, L., Sato, A., Karow, M., Adams, N.C., Gale, N.W., Iwasaki, A., & Flavell, R.A. (2004) Proc. Natl. Acad. Sci. USA, 101, 5598–5603.
15) Heil, F., Hemmi, H., Hochrein, H., Ampenberger, F., Kirschning, C., Akira, S., Lipford, G., Wagner, H., & Bauer, S. (2004) Science, 303, 1526–1529.
16) Diebold, S.S., Kaisho, T., Hemmi, H., Akira, S., & Sousa, C.R.E. (2004) Science, 303, 1529–1531.
17) Hemmi, H., Takeuchi, O., Kawai, T., Kaisho, T., Sato, S., Sanjo, H., Matsumoto, M., Hoshino, K., Wagner, H., Takeda, K., & Akira, S. (2000) Nature, 408, 740–745.
18) Matsushima, N., Tanaka, T., Enkhbayar, P., Mikami, T., Taga, M., Yamada, K., & Kuroki, Y. (2007) BMC Genomics, 8, 124.
19) Bell, J.K., Mullen, G.E., Leifer, C.A., Mazzoni, A., Davies, D.R., & Segal, D.M. (2003) Trends Immunol., 24, 528–533.
20) Song, D.H. & Lee, J.O. (2012) Immunol. Rev., 250, 216–229.
21) Nagai, Y., Akashi, S., Nagafuku, M., Ogata, M., Iwakura, Y., Akira, S., Kitamura, T., Kosugi, A., Kimoto, M., & Miyake, K. (2002) Nat. Immunol., 3, 667–672.
22) Ohto, U., Fukase, K., Miyake, K., & Satow, Y. (2007) Science, 316, 1632–1634.
23) Kim, H.M., Park, B.S., Kim, J.I., Kim, S.E., Lee, J., Oh, S.C., Enkhbayar, P., Matsushima, N., Lee, H., Yoo, O.J., & Lee, J.O. (2007) Cell, 130, 906–917.
24) Park, B.S., Song, D.H., Kim, H.M., Choi, B.S., Lee, H., & Lee, J.O. (2009) Nature, 458, 1191–1195.
25) Ohto, U., Fukase, K., Miyake, K., & Shimizu, T. (2012) Proc. Natl. Acad. Sci. USA, 109, 7421–7426.
26) Raetz, C.R.H., Reynolds, C.M., Trent, M.S., & Bishop, R.E. (2007) Annu. Rev. Biochem., 76, 295–329.
27) Golenbock, D.T., Hampton, R.Y., Qureshi, N., Takayama, K., & Raetz, C.R.H. (1991) J. Biol. Chem., 266, 19490–19498.
28) Meng, J.M., Drolet, J.R., Monks, B.G., & Golenbock, D.T. (2010) J. Biol. Chem., 285, 27935–27943.
29) Meng, J., Lien, E., & Golenbock, D.T. (2010) J. Biol. Chem., 285, 8695–8702.
30) Vasl, J., Oblak, A., Gioannini, T.L., Weiss, J.P., & Jerala, R. (2009) J. Immunol., 183, 5138–5145.
31) Walsh, C., Gangloff, M., Monie, T., Smyth, T., Wei, B., McKinley, T.J., Maskell, D., Gay, N., & Bryant, C. (2008) J. Immunol., 181, 1245–1254.
32) Muroi, M. & Tanamoto, K. (2006) J. Biol. Chem., 281, 5484–5491.
33) Saitoh, S., Akashi, S., Yamada, T., Tanimura, N., Kobayashi, M., Konno, K., Matsumoto, F., Fukase, K., Kusumoto, S., Nagai, Y., Kusumoto, Y., Kosugi, A., & Miyake, K. (2004) Int. Immunol., 16, 961–969.
34) Hajjar, A.M., Ernst, R.K., Tsai, J.H., Wilson, C.B., & Miller, S.I. (2002) Nat. Immunol., 3, 354–359.
35) Jurk, M., Heil, F., Vollmer, J., Schetter, C., Krieg, A.M., Wagner, H., Lipford, G., & Bauer, S. (2002) Nat. Immunol., 3, 499–499.
36) Hemmi, H., Kaisho, T., Takeuchi, O., Sato, S., Sanjo, H., Hoshino, K., Horiuchi, T., Tomizawa, H., Takeda, K., & Akira, S. (2002) Nat. Immunol., 3, 196–200.
37) Ishii, N., Funami, K., Tatematsu, M., Seya, T., & Matsumoto, M. (2014) J. Immunol., 193, 5118–5128.
38) Ewald, S.E., Engel, A., Lee, J., Wang, M.Q., Bogyo, M., & Barton, G.M. (2011) J. Exp. Med., 208, 643–651.
39) Sepulveda, F.E., Maschalidi, S., Colisson, R., Heslop, L., Ghirelli, C., Sakka, E., Lennon-Dumenil, A.M., Amigorena, S., Cabanie, L., & Manoury, B. (2009) Immunity, 31, 737–748.
40) Park, B., Brinkmann, M.M., Spooner, E., Lee, C.C., Kim, Y.M., & Ploegh, H.L. (2008) Nat. Immunol., 9, 1407–1414.
41) Ewald, S.E., Lee, B.L., Lau, L., Wickliffe, K.E., Shi, G.P., Chapman, H.A., & Barton, G.M. (2008) Nature, 456, 658–688.
42) Tanji, H., Ohto, U., Shibata, T., Miyake, K., & Shimizu, T. (2013) Science, 339, 1426–1429.
43) Tanji, H., Ohto, U., Shibata, T., Taoka, M., Yamauchi, Y., Isobe, T., Miyake, K., & Shimizu, T. (2015) Nat. Struct. Mol. Biol., 22, 109–115.
44) Krieg, A.M., Yi, A.K., Matson, S., Waldschmidt, T.J., Bishop, G.A., Teasdale, R., Koretzky, G.A., & Klinman, D.M. (1995) Nature, 374, 546–549.
45) Ohto, U., Shibata, T., Tanji, H., Ishida, H., Krayukhina, E., Uchiyama, S., Miyake, K., & Shimizu, T. (2015) Nature, 520, 702–705.
46) Tanji, H., Ohto, U., Motoi, Y., Shibata, T., Miyake, K., & Shimizu, T. (2016) Proc. Natl. Acad. Sci. USA, 113, 3012–3017.
47) Leonard, J.N., Ghirlando, R., Askins, J., Bell, J.K., Margulies, D.H., Davies, D.R., & Segal, D.M. (2008) Proc. Natl. Acad. Sci. USA, 105, 258–263.
48) Bell, J.K., Botos, I., Hall, P.R., Askins, J., Shiloach, J., Segal, D.M., & Davies, D.R. (2005) Proc. Natl. Acad. Sci. USA, 102, 10976–10980.
著者紹介Author Profile
 大戸 梅治(おおと うめはる)
大戸 梅治(おおと うめはる)東京大学大学院薬学系研究科准教授.薬学博士.
略歴1978年宮崎県に生る.2002年東京大学薬学部卒業.07年同大学院薬学系研究科博士課程修了.同年東京大学大学院薬学系研究科助教.13年同講師.16年より現職.
研究テーマと抱負受容体の活性制御機構を明らかにする.できることを確実に進める.
ウェブサイトhttp://www.f.u-tokyo.ac.jp/~kouzou/index.html
趣味ランニング.