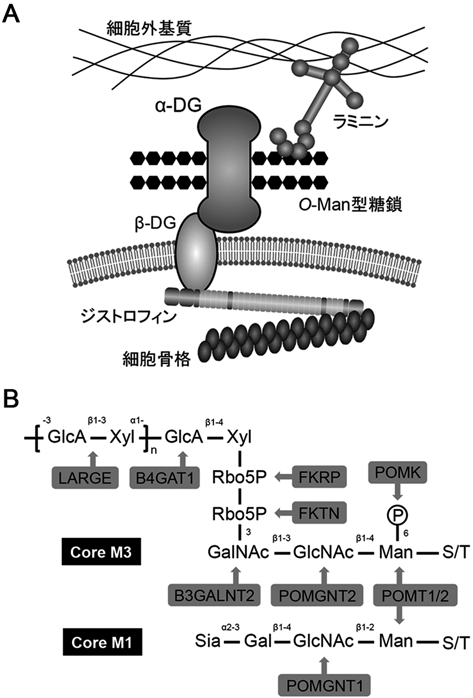
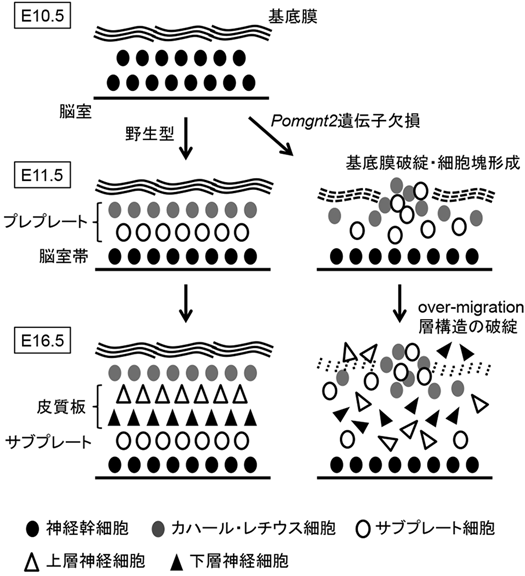
ジストログリカン糖鎖の生合成機構と大脳皮質形成における役割Dystroglycan glycosylation: structure, synthetic pathway, and in vivo role in the brain development
京都大学大学院医学研究科人間健康科学系専攻Department of Biological Chemistry, Human Health Sciences, Graduate School of Medicine, Kyoto University ◇ 〒606–8507 京都府京都市左京区聖護院川原町53 ◇ 53 Kawahara-cho, Shogoin, Sakyo-ku, Kyoto 606–8507, Japan
発行日:2016年8月25日Published: August 25, 2016