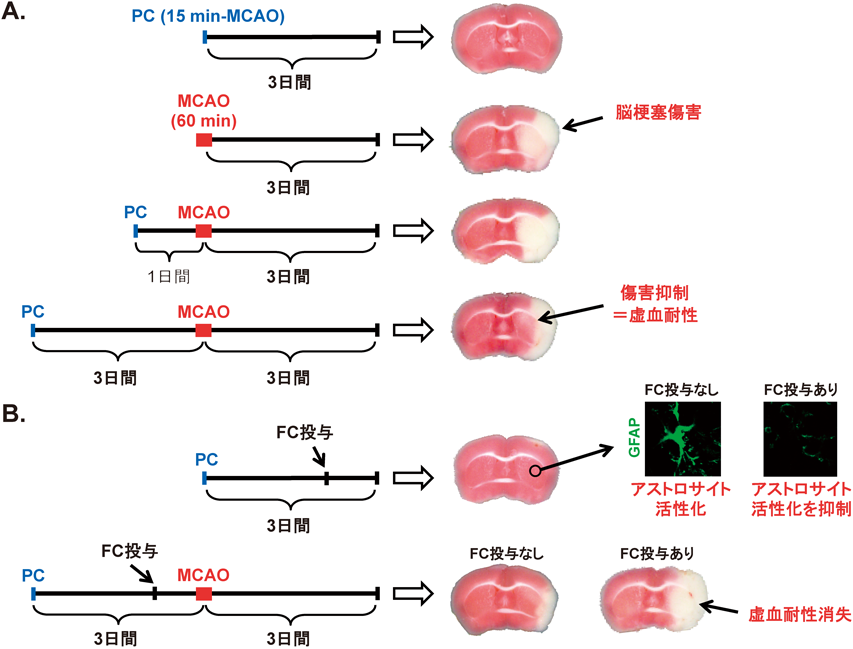
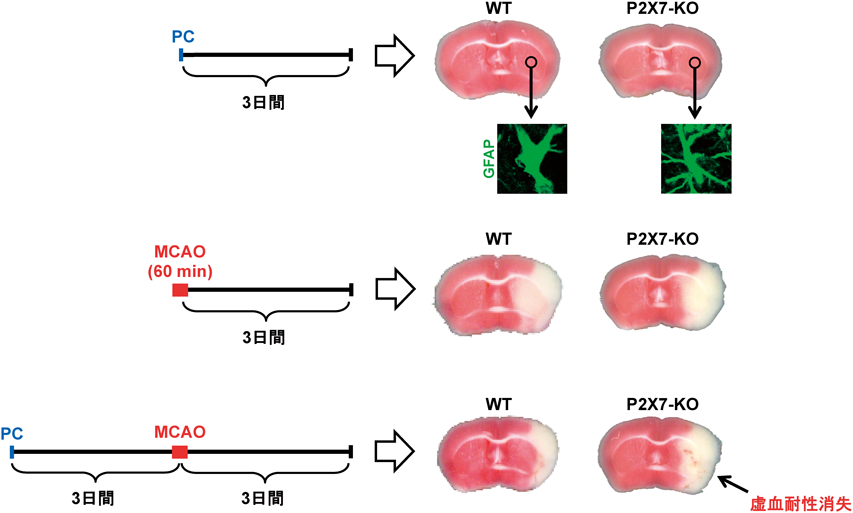
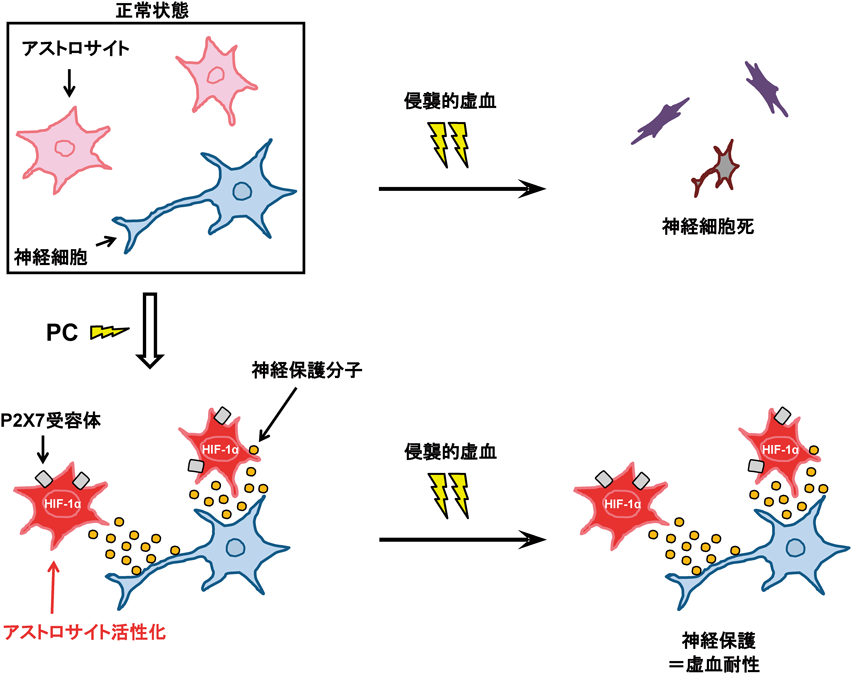
グリア細胞によって誘導される虚血耐性Glia-mediated ischemic tolerance
1 山梨大学大学院総合研究部医学域薬理学Department of Neuropharmacology, Interdisciplinary Graduate School of Medicine, University of Yamanashi ◇ 〒409–3898 山梨県中央市下河東1110 ◇ 1110 Shimokato, Chuo, Yamanashi 409–3898, Japan
2 山梨大学医学部リエゾンアカデミーDepartment of Liaison Academy, Faculty of Medicine, University of Yamanashi ◇ 〒409–3898 山梨県中央市下河東1110 ◇ 1110 Shimokato, Chuo, Yamanashi 409–3898, Japan
発行日:2016年8月25日Published: August 25, 2016