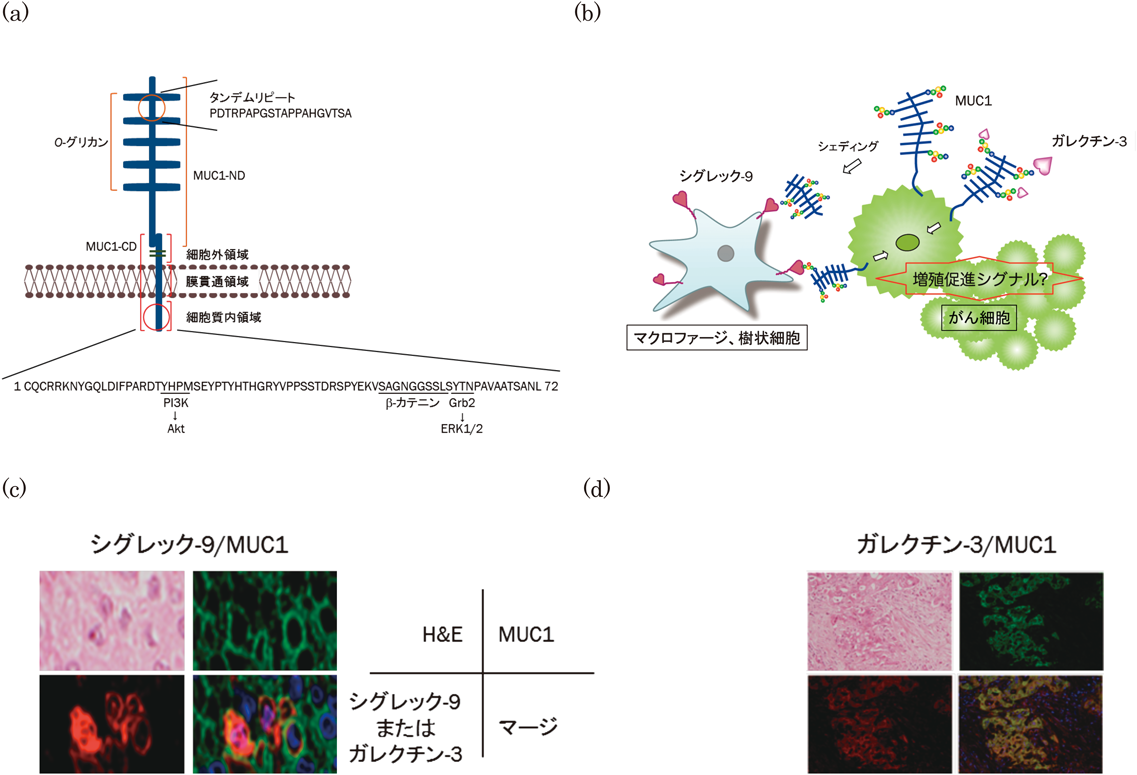
ムチンのコアタンパク質は,現在,遺伝子として20数種が同定され,大きく分泌型と膜結合型に分類される.前者は主にゲルを形成して粘膜層を構成し,保護作用などを担う.後者に属するムチンであるムチン1(MUC1)は上皮細胞に普遍的に発現し,上皮性がんにおいて発現量が増加する.MUC1は1本のポリペプチド鎖として合成された後,自己切断によって,大半の細胞外領域を形成するN末端ドメイン(MUC1-ND)と短い細胞外領域,膜貫通領域,細胞質側領域で構成されるC末端ドメイン(MUC-CD)が非共有結合した状態で細胞表面に発現する(図1a).MUC1-NDは,主に反復する一定のアミノ酸配列(タンデムリピート)で構成され,セリン,トレオニンに富み,多数のO-グリカンの結合部位となる.MUC1-CDの細胞質側はアミノ酸72個のポリペプチド鎖からなり,複数の情報伝達に関連する分子との相互作用を通じて,がん細胞の増殖や浸潤能の促進,アポトーシスの抑制などに関与するとされている.
がんの大半は上皮細胞由来であるが,がん化に伴うムチンの変化については,一般的に物質的な変化と存在状態の変化があげられる.前者は発現量の増加と糖鎖の変化である.糖鎖の変化はがん関連糖鎖抗原の発現として多くの研究がなされてきた1–3).後者はがん化に伴う上皮組織の極性の消失に起因する変化である.正常上皮組織では,ムチンは細胞の頂端側に輸送されるが,極性の消失後は,細胞表層全体に輸送される.すなわち,上皮–間葉系転換後に形成されるがん組織微小環境において,がん細胞上のMUC1と間質細胞上の表層タンパク質や,各種細胞が産生する可溶性因子,とりわけレクチンとの相互作用が想定される(図1b).
2. 増殖因子を起点としたMUC1やMUC4仲介性の情報伝達4, 5)
従来,MUC1の関与する情報伝達については,細胞膜上でのMUC1と増殖因子受容体との相互作用により,増殖因子を起点としたシグナルをMUC1が中継する機構が明らかにされてきた6).図1aにシグナル伝達に関与する介在因子,あるいは転写因子のリクルートされる部位(図1aの配列下線部分)を示す.その中でMUC1とErbB受容体の相互作用によるErbBシグナルの増強はERK経路などを活性化する.また,繊維芽細胞増殖因子(FGF)を起点として,FGF受容体3とMUC1の相互作用はMUC1の細胞質部分のリン酸化に続いて,β-カテニンのリクルートと核移行を促進する7).さらに,エストロゲン受容体α(ERα)もMUC1と複合体を形成し,ERα DNA結合ドメインへの結合によりエストロゲン仲介の細胞増殖を亢進する.MUC4についても,その上皮増殖因子(EGF)様ドメインを介してErbB受容体と相互作用するとされている8).ErbB3はリガンドであるニューレグリンと結合するとErbB2と複合体を形成する.MUC4-ErbB2/ErbB3複合体の形成は受容体を安定化するとともにErbB2のリン酸化を亢進し,AktやERK経路を活性化する.
3. レクチンの結合に伴うMUC1を介した情報伝達と腫瘍悪性化
我々は,多様でかつ多数の糖鎖を発現するMUC1ががん組織微小環境に存在することが想定されるレクチンと結合した場合のシグナル伝達について検討してきた.
1)がん組織微小環境におけるシグレック-9,ガレクチン-3およびMUC1の分布
マクロファージなどの免疫系細胞の細胞表面には,シグレックファミリーなどの多くのレクチンが発現している.シグレックファミリーはシアログリカンを認識することから,多様なシアロO-グリカンを発現するMUC1は好適なカウンターリセプターになることが予想された.組換え体を用いた結合実験により,マクロファージなどに発現しているシグレックファミリーの中で,シグレック-9が比較的強い結合活性を持つことがわかった.一方,可溶性レクチンとしては上皮性がん患者において血中濃度が上昇し,予後不良因子とされ,また,MUC1の内在性リガンドであるとされているガレクチン-3を取り上げた9, 10).
ヒトがん組織標本(膵臓がん,胃がん,乳がん,大腸がん)を用いて,MUC1,シグレック-9,およびガレクチン-3の分布を調べた.図1cには大腸がん組織の例を示す.シグレック-9については,MUC1陽性細胞の中に浸潤したシグレック-9陽性細胞が点在し,両分子が相互作用する可能性が十分に予想された.また,ガレクチン-3についてはMUC1陽性細胞の分布と一致した(図1d).
2)シグレック-9やガレクチン-3のMUC1への結合に伴うシグナル伝達と腫瘍の悪性化11–13)
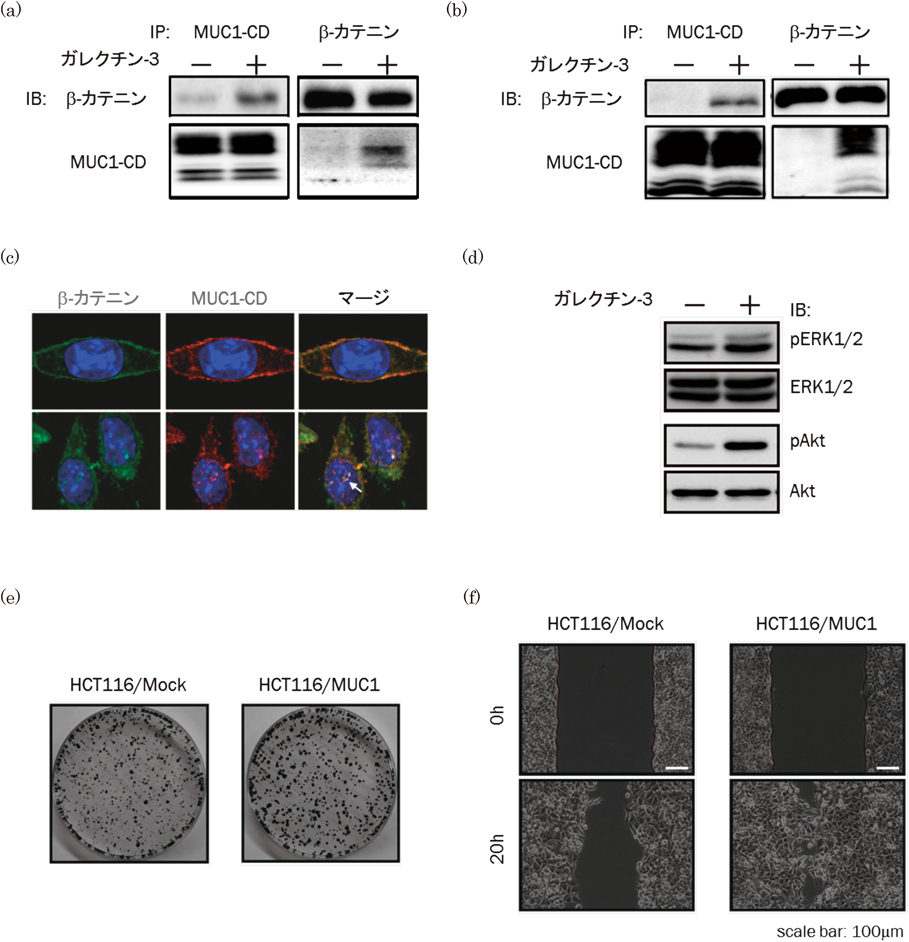
我々はシグレック-9やガレクチン-3がMUC1の糖鎖に結合し,直接シグナル伝達が惹起される可能性について検討した.細胞としては,ヒト大腸がん由来HCT116細胞,ヒト肺がん由来A549細胞,マウス繊維芽細胞3T3細胞にMUC1を強制発現したそれぞれHCT116/MUC1, A549/MUC1,マウス3T3/MUC1細胞およびそれぞれのコントロール細胞,さらに内在性のMUC1を発現するヒト卵巣がん由来SKOV3細胞(SKOV3/Scr)と同細胞のMUC1をノックダウンした細胞(SKOV3/Sci)などを用いた.細胞の培養液に可溶型シグレック-9組換え体あるいはガレクチン-3を加え,細胞を刺激後,可溶化した.この細胞抽出液(ライセート)よりMUC1-CDあるいはβ-カテニンに対する抗体による免疫沈降物を電気泳動後,ウエスタンブロットを行った.MUC1-CDおよびβ-カテニンの沈降物中にはそれぞれβ-カテニンとMUC1-CDが認められた(図2a, b).リクルートされたβ-カテニンは,レクチンの濃度依存的に増加し,細胞の刺激後20~30分でピークとなった.HCT116/MUC1細胞に可溶型シグレック-9を作用させ,1時間後の細胞について,免疫染色によりβ-カテニンの分布を見たところ,核内に集積するβ-カテニンが観察され,かつMUC1-CDとの共局在が認められた(図2c).したがって,MUC1-CDとβ-カテニンは複合体を形成して核内に輸送されたものと考えられる.また,実際のがん組織微小環境におけるシグレック-9については,免疫細胞上に発現するシグレック-9のカウンターリセプターとしてがん細胞上のMUC1が機能すると考えられ,この環境をミミックする系として,シグレック-9強制発現細胞株HEK293/Sig9およびMock細胞を作製し,MUC1発現細胞との共培養による効果を測定した.Mock細胞と比較して,HEK293/Sig9細胞との共培養により,MUC1-CDへのβ-カテニンのリクルートは6~7倍増加した.したがって,シグレック-9とMUC1による免疫細胞とがん細胞の細胞間相互作用によって,腫瘍の増殖が促進されているものと考えられる.
次に,図1aに示すように,MUC1の細胞質側のペプチド鎖中にPI3Kp85とGrb2の結合部位も存在することから,ERK1/2およびAktのリン酸化についても検討した.種々の細胞において,ガレクチン-3の刺激によるERK1/2およびAktのリン酸化レベルの変化は,MUC1の強制発現によって上昇し(図2d),ノックダウンによって減少した.本反応は,EGF受容体阻害剤存在下でも同様に検出され,前述したEGFなどを起点とした情報伝達とは異なることが確認された.次に,これらの情報伝達の結果としての生物学的効果を測定した.足場依存性コロニーの形成(図2e)およびwound healing assayによる移動能の測定(図2f)では,いずれもMUC1の発現によってその効果が亢進した.HCT116細胞のみならず,A549細胞およびSKOV3細胞について,MUC1の強制発現あるいはノックダウンによるMUC1のレベルの違いに応じて増殖能や移動能が変化した.さらに,これらの細胞の内在性のガレクチン-3をノックダウンすると増殖能および移動能は低下し,培養液にガレクチン-3を加えると回復した.これらの結果より,これらの形質はガレクチン-3とMUC1の相互作用がもたらした現象であることが確認された.
3)MUC1によるuPAの誘導14)
HCT116/MUC1とHCT116/Mock細胞のmRNAの発現をDNAマイクロアレイにより比較したところ,MUC1発現細胞においてウロキナーゼ型プラスミノーゲン活性化因子(urokinase-type plasminogen activator:uPA)mRNAの著しい発現の増加が認められた.また,前項で述べたさまざまな細胞を用い,タンパク質レベルでuPAの発現を検討したところMUC1のレベルに対応してuPAのレベルが増減した.また,種々のヒトがん組織におけるMUC1およびuPAの分布も一致した.また,細胞のライセートよりNF-κBとMUC1-CDが免疫沈降によって共沈することから,両分子は複合体を形成していることがわかった(図3a).この結果は,uPAの転写因子の一つとしてNF-κBが同定されていることおよびMUC1の細胞質側のペプチド鎖上にNF-κBの結合部位が存在するという報告と符合した15).また,この複合体は核内に検出され,その存在量はMUC1の発現量に呼応した.したがって,MUC1の発現レベルの上昇はMUC1-CD/NF-κB複合体の形成と核移行を促進することがわかった.
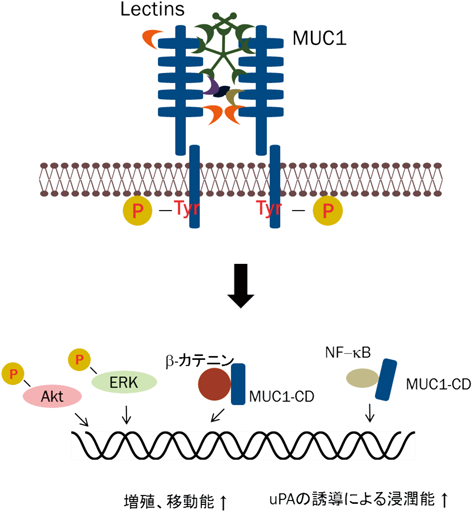
次に,ChIPアッセイによってMUC1-CD/NF-κB複合体のuPA転写制御への関与を検討した.クロマチン画分からのNF-κB免疫沈降物中のDNAを増幅するとMock細胞に比してHCT116/MUC1では約4倍の該当するDNAが増幅された(図3b).さらに,抗MUC1-CD抗体を用いたreChIPアッセイによってもMUC1-CD/NF-κB複合体がuPAプロモーター領域に結合していることが確認された.次に,ルシフェラーゼアッセイによってuPA転写活性を測定するとMUC1の発現レベルに応じて増減した.また,uPAプロモーターのNF-κB結合領域に変異を導入したDNAを用いたルシフェラーゼアッセイでは,MUC1発現細胞にみられた転写活性がコントロール細胞のレベルまで減少した.これらの結果は,MUC1-CD/NF-κB複合体によってuPAの転写が制御されていることを示している.分泌されたuPAをELISAで測定するとHCT116細胞では,MUC1の強制発現によって約10倍増加した(図3c).また,培養液中のuPAおよびMMP2/9の活性化型をザイモグラフィーで測定するとMUC1のレベルに応じて活性型が増減した.Boyden chamberを用いて,細胞の浸潤能を測定するとHCT116細胞ではMUC1の強制発現に伴い浸潤能は約2倍となった.浸潤能を指標として,JSH-23(NF-κB阻害剤),GM6001(MMPs阻害剤),アミロライド(uPA阻害剤)の効果を調べたところ,いずれの阻害剤によっても約60%の浸潤能の阻害がみられた.したがって,浸潤能の亢進に至るプロセスについて明らかにしてきた一連の反応が寄与しているものと考えられる.なお,MUC1によるuPAの誘導についても,MUC1へのガレクチン-3の結合によって亢進することがわかった(未発表データ).このように,従来の増殖因子を起点として,シグナルを中継するMUC1の機能に加えて,レクチンをリガンドとして腫瘍悪性化を担うMUC1の新規な機能が明らかとなった.一連の研究で明らかにしたMUC1を介した様々なシグナル伝達の模式図を図4に示す.
引用文献References
1) Brockhausen, I. (2006) EMBO Rep., 7, 599–604.
2) 中田博,山科郁男(1994)生化学,60, 1387–1400.
3) Wopereis, S., Lefeber, D.J., Morava, E., & Wevers, R.A. (2006) Clin. Chem., 52, 574–600.
4) Singh, P.K. & Hollingsworth, M.A. (2006) Trends Cell Biol., 16, 467–476.
5) Bafna, S., Kaur, S., & Batra, S.K. (2010) Oncogene, 29, 2893–2904.
6) Kufe, D.W. (2013) Oncogene, 32, 1073–1081.
7) Ren, J., Raina, D., Chen, W., Li, G., Huang, L., & Kufe, D.W. (2006) Mol. Cancer Res., 4, 873–883.
8) Carraway, K.L., Funes, M., Workman, H.C., & Sweeney, C. (2007) Curr. Top. Dev. Biol., 4, 873–883.
9) Iurisci, I., Tinari, N., Natoli, C., Angelucci, D., Cianchetti, E., & Lacobelli, S. (2000) Clin. Cancer Res., 6, 1389–1393.
10) Yu, L.G., Andrews, N., Zhao, Q., McKean, D., Williams, J.F., Connor, L.J., Gerasimenko, O.V., Hilkens, J., Hirabayashi, J., Kasai, K., & Rhodes, J.M. (2007) J. Biol. Chem., 282, 773–781.
11) Tanida, S., Akita, K., Ishida, A., Mori, Y., Toda, M., Inoue, M., Ohta, M., Yashiro, M., Sawada, T., Hirakawa, K., & Nakada, H. (2013) J. Biol. Chem., 288, 31842–31852.
12) Tanida, S., Mori, Y., Ishida, A., Akita, K., & Nakada, H. (2014) Biochim. Biophys. Acta, 1840, 1790–1797.
13) Mori, Y., Akita, K., Yashiro, M., Sawada, T., Hirakawa, K., Murata, T., & Nakada, H. (2015) J. Biol. Chem., 290, 26125–26140.
14) Mori, Y., Akita, K., Tanida, S., Ishida, A., Toda, M., Inoue, M., Yashiro, M., Sawada, T., Hirakawa, K., & Nakada, H. (2014) J. Biol. Chem., 289, 35193–35204.
15) Ahmad, R., Raina, D., Joshi, M.D., Kawano, T., Ren, J., Kharbanda, S., & Kufe, D. (2009) Cancer Res., 69, 7013–7021.